


Tomography 2024, 10(5), 654-659; https://doi.org/10.3390/tomography10050050 (registering DOI) - 25 Apr 2024
Abstract
This brief report aimed to show the utility of photon-counting technology alongside standard cranial imaging protocols for visualizing shunt valves in a patient’s cranial computed tomography scan. Photon-counting CT scans with cranial protocols were retrospectively surveyed and four types of shunt valves were
[...] Read more.
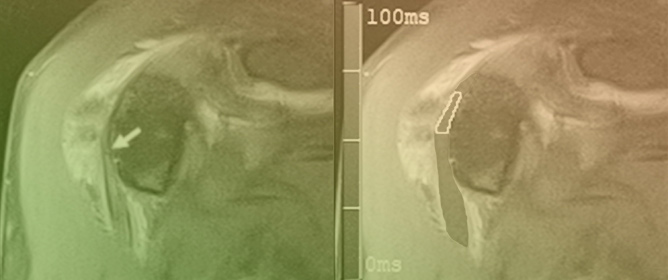
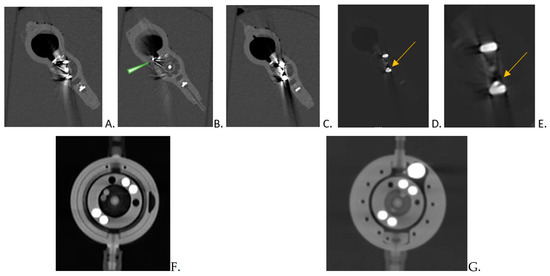
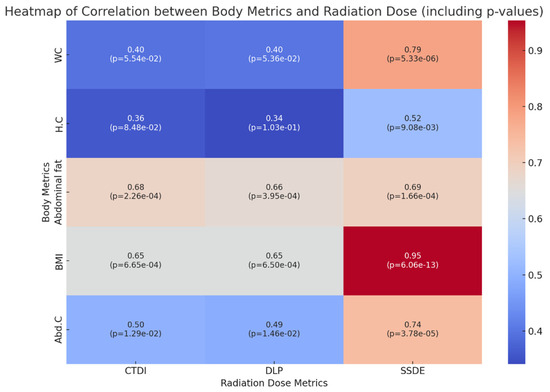
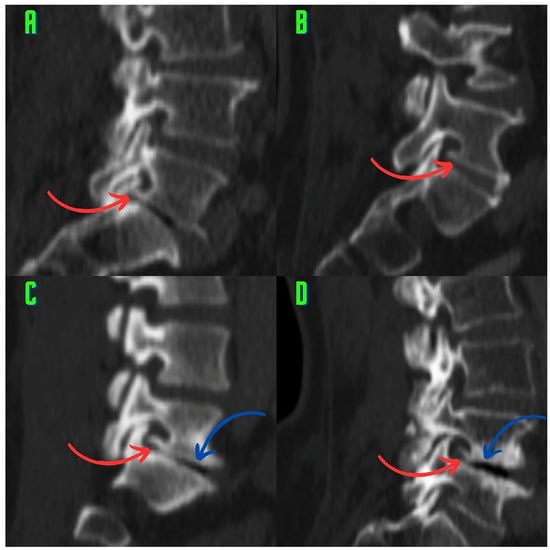
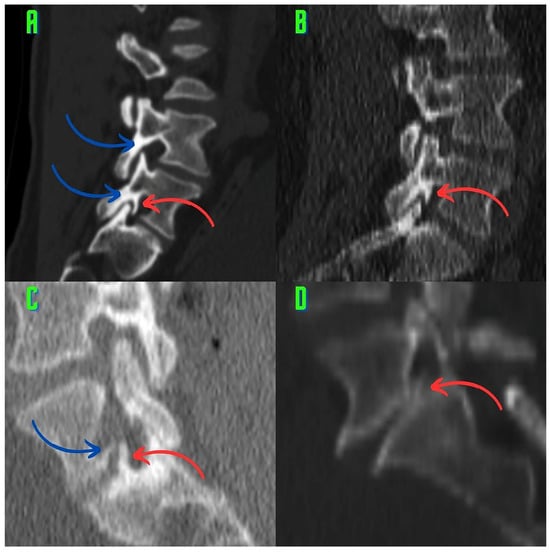
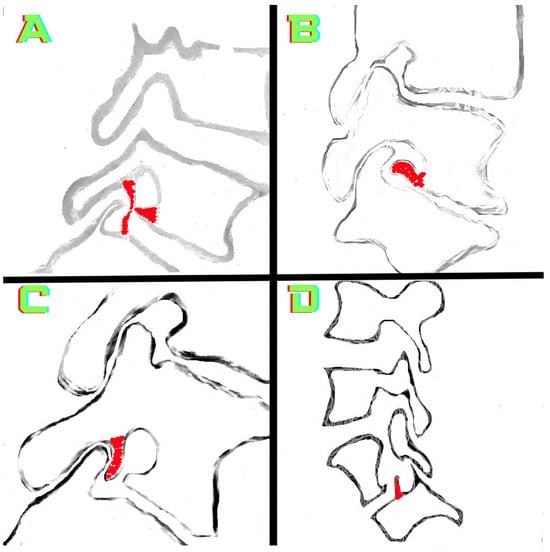
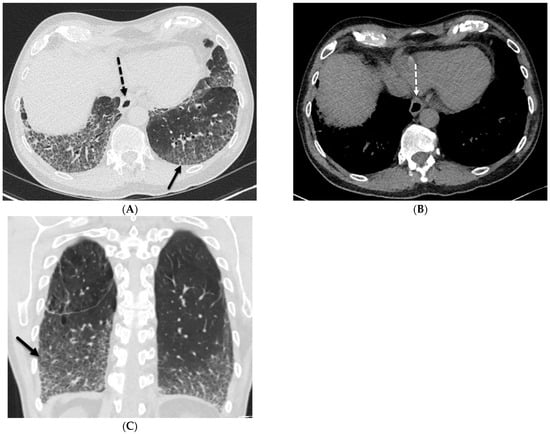
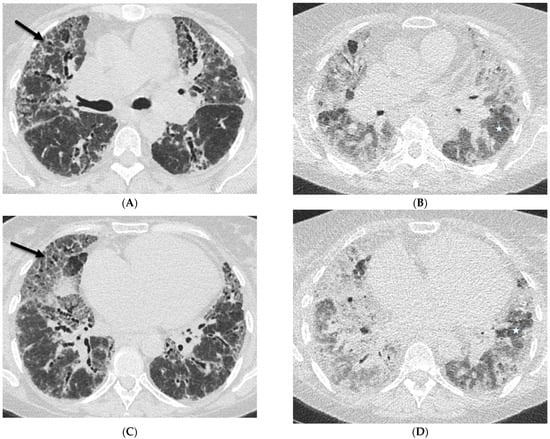
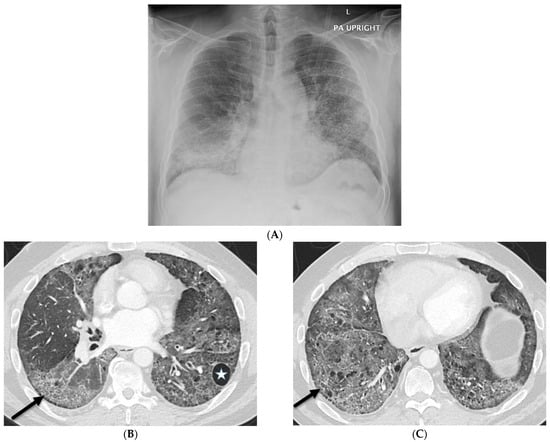
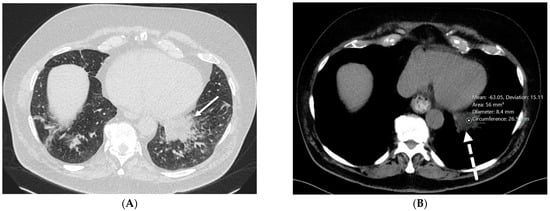
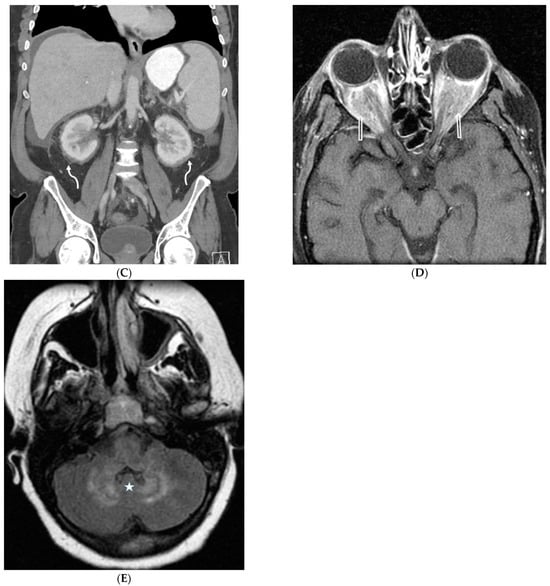
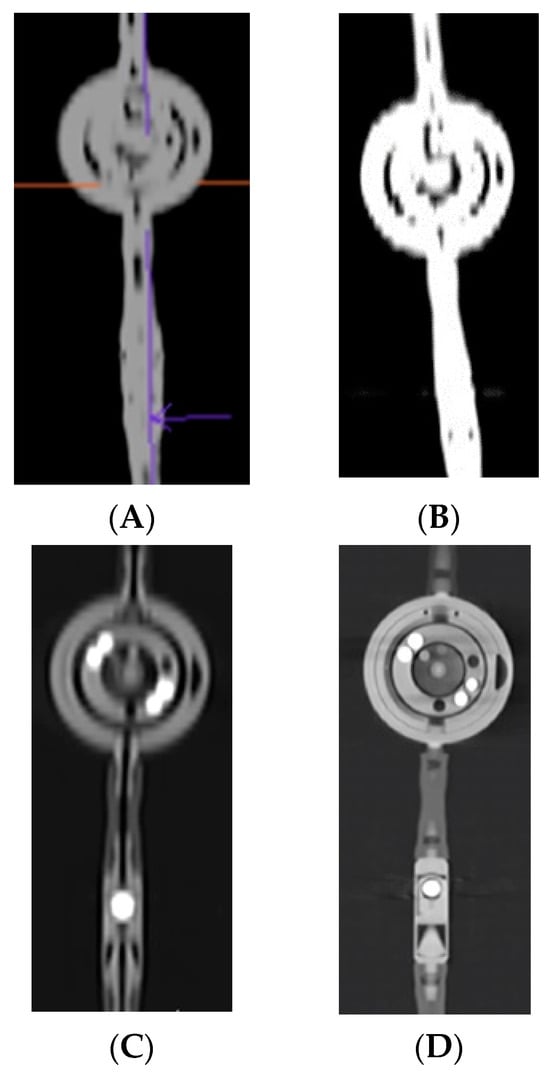
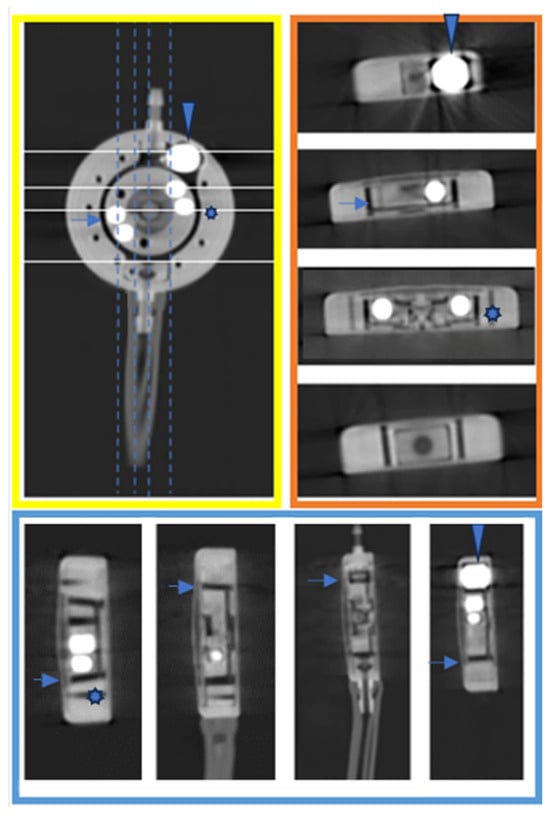
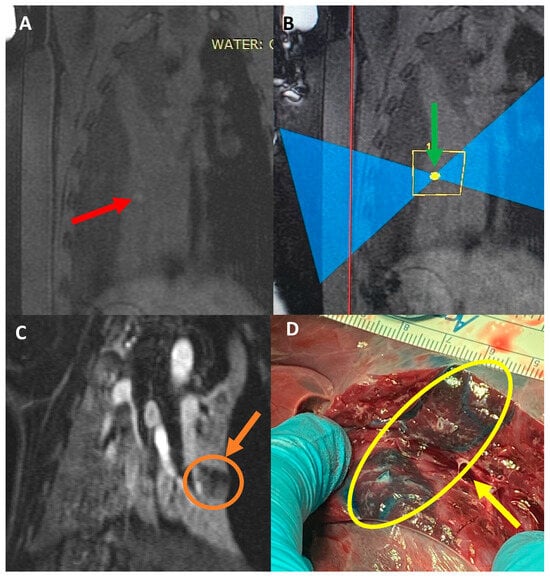
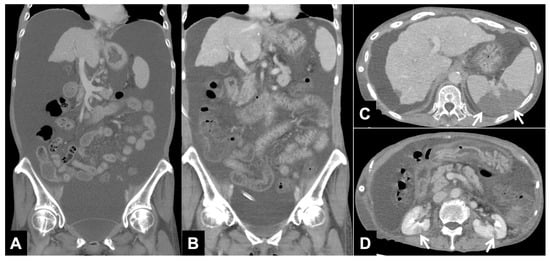
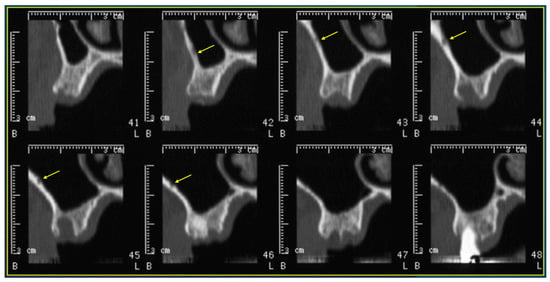
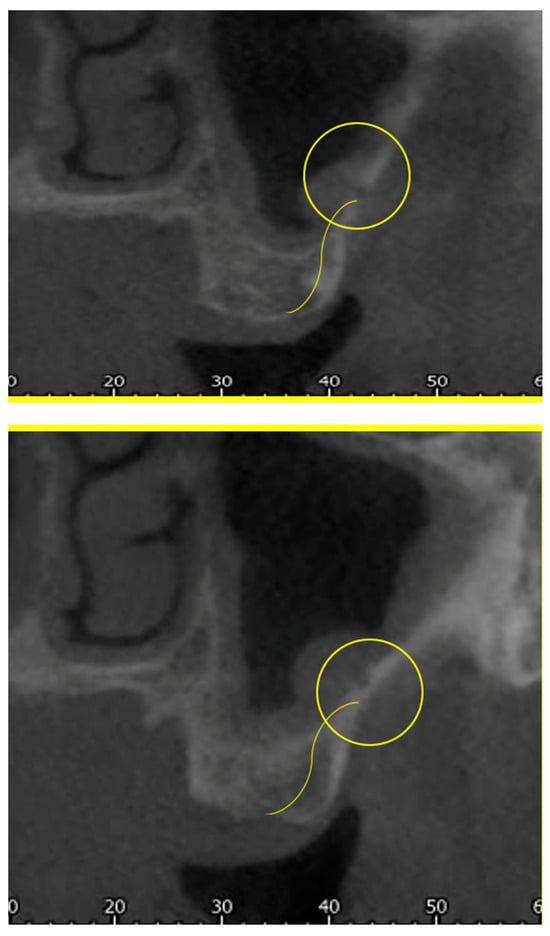
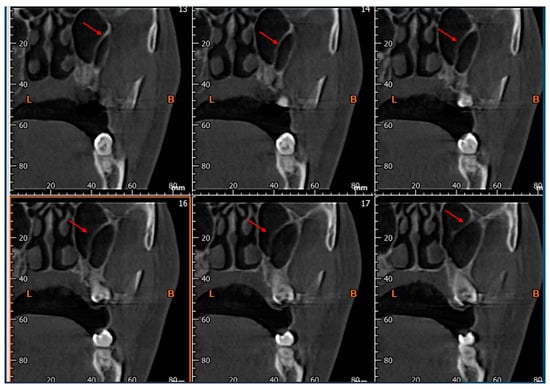
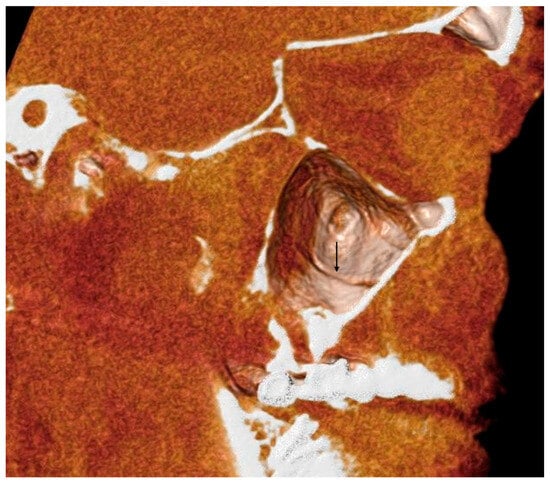
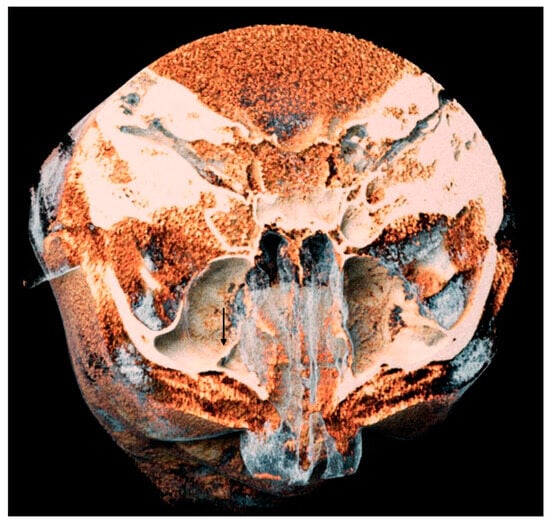
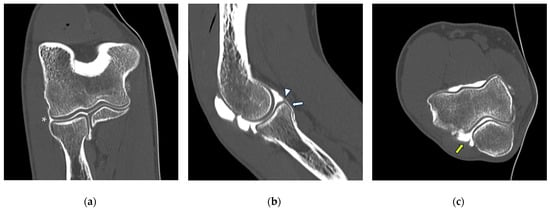
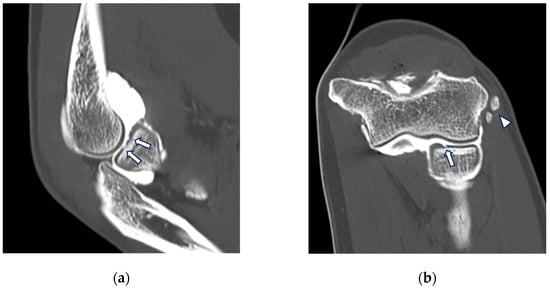
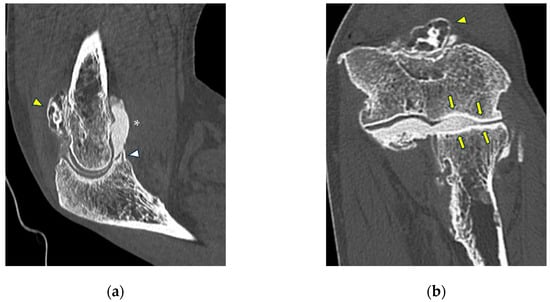
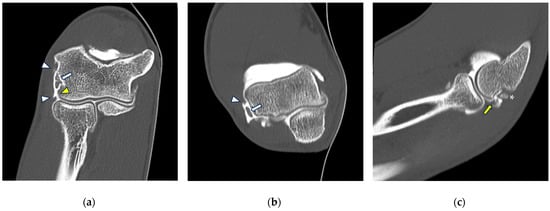
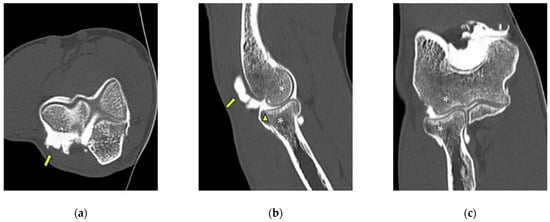
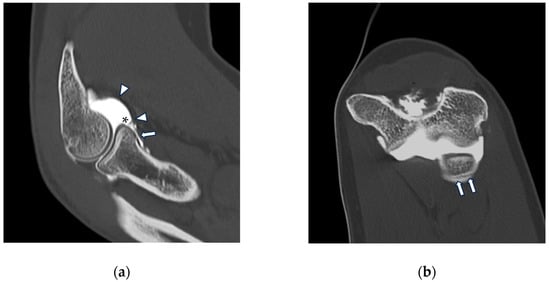
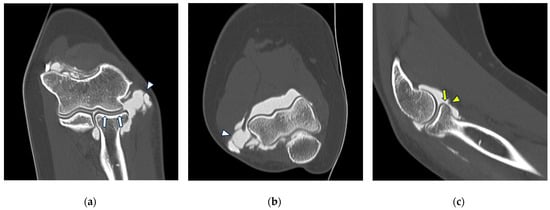
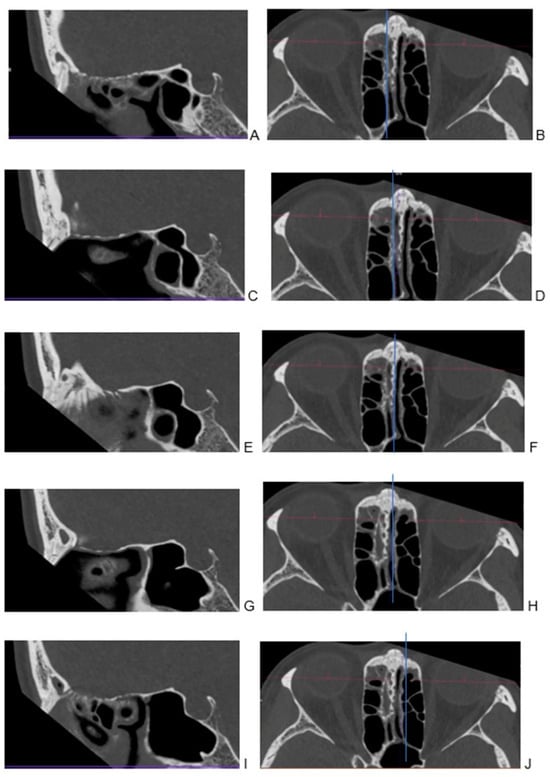
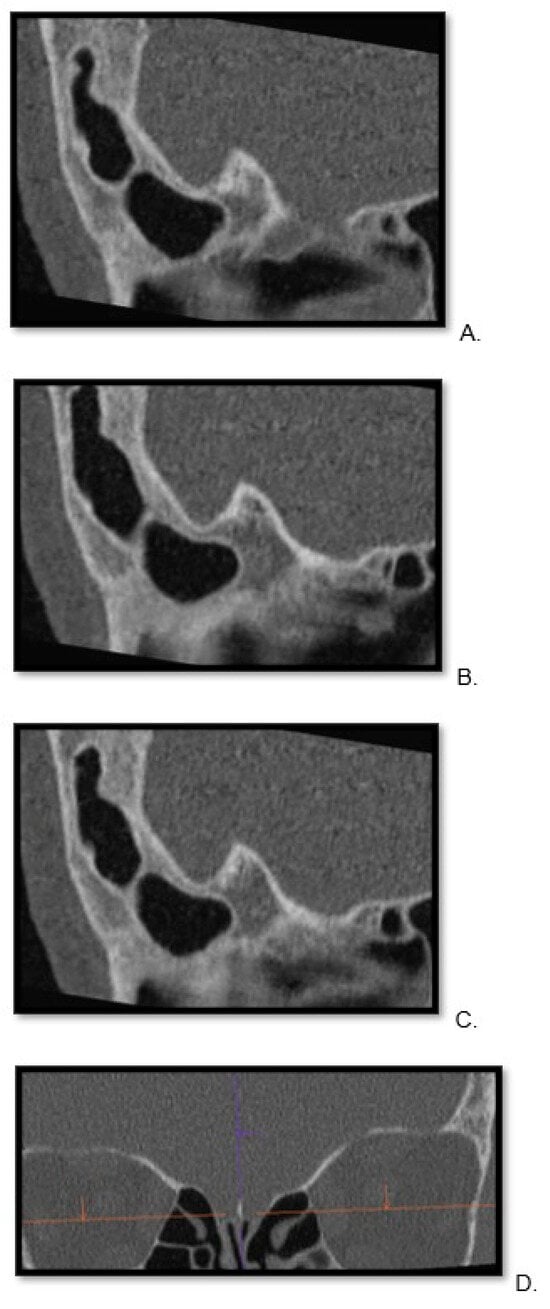
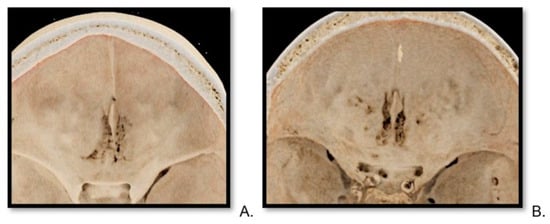
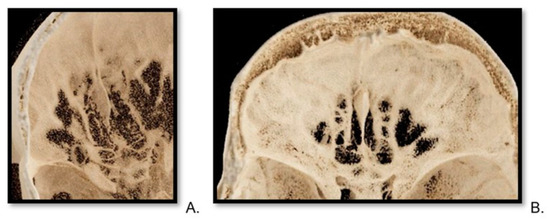
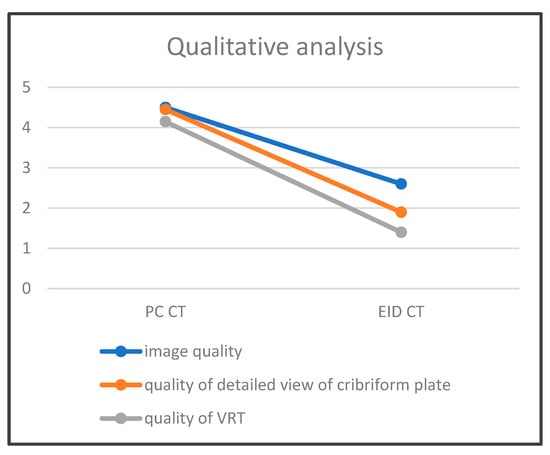
This brief report aimed to show the utility of photon-counting technology alongside standard cranial imaging protocols for visualizing shunt valves in a patient’s cranial computed tomography scan. Photon-counting CT scans with cranial protocols were retrospectively surveyed and four types of shunt valves were encountered: proGAV 2.0®, M.blue®, Codman Certas®, and proSA®. These scans were compared with those obtained from non-photon-counting scanners at different time points for the same patients. The analysis of these findings demonstrated the usefulness of photon-counting technology for the clear and precise visualization of shunt valves without any additional radiation or special reconstruction patterns. The enhanced utility of photon-counting is highlighted by providing superior spatial resolution compared to other CT detectors. This technology facilitates a more accurate characterization of shunt valves and may support the detection of subtle abnormalities and a precise assessment of shunt valves.
Full article
(This article belongs to the Section Neuroimaging)
►
Show Figures

Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}