


Tomography 2024, 10(4), 574-608; https://doi.org/10.3390/tomography10040045 - 17 Apr 2024
Abstract
►
Show Figures
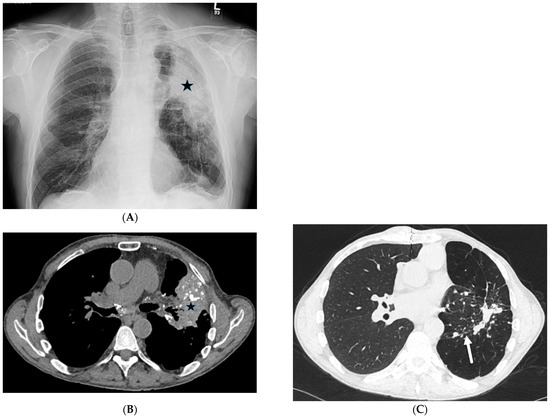
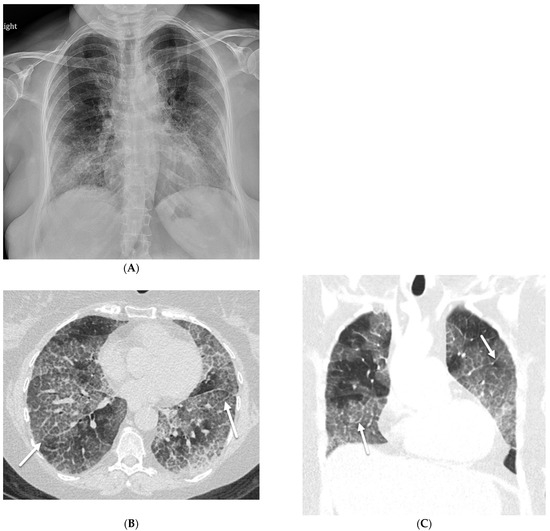
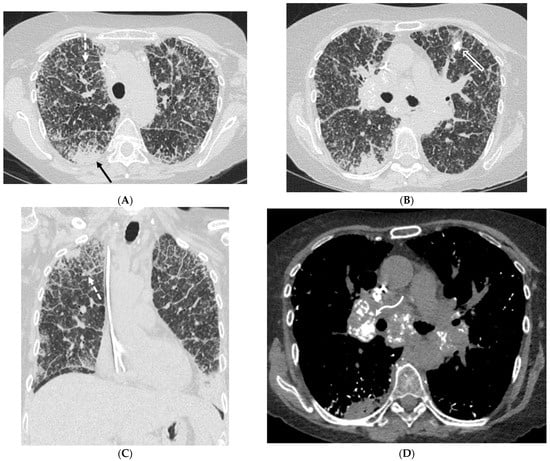
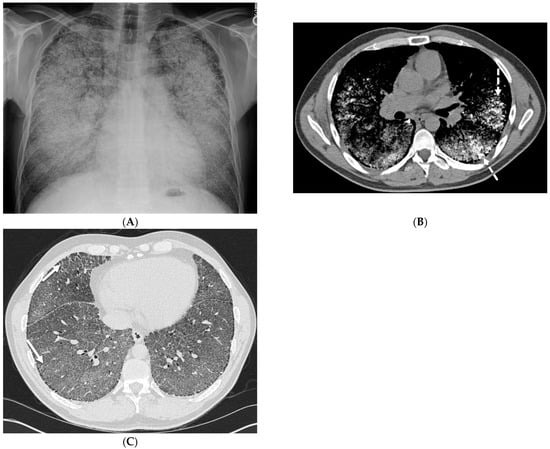
Interlobular septa thickening (ILST) is a common and easily recognized feature on computed tomography (CT) images in many lung disorders. ILST thickening can be smooth (most common), nodular, or irregular. Smooth ILST can be seen in pulmonary edema, pulmonary alveolar proteinosis, and lymphangitic
[...] Read more.
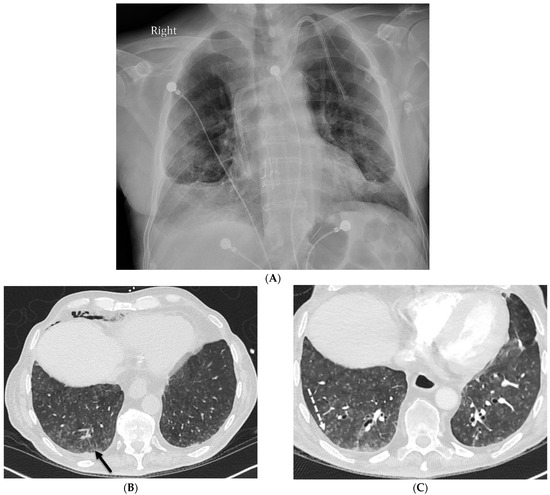
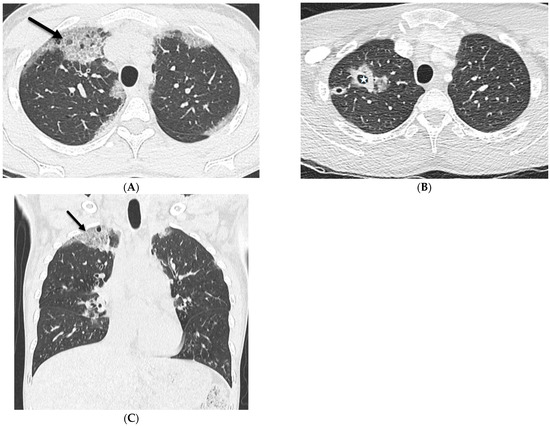
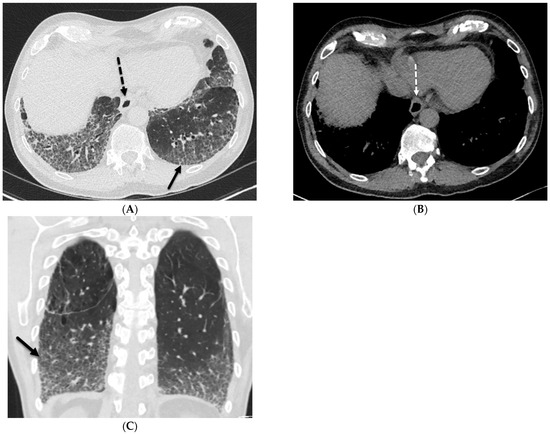
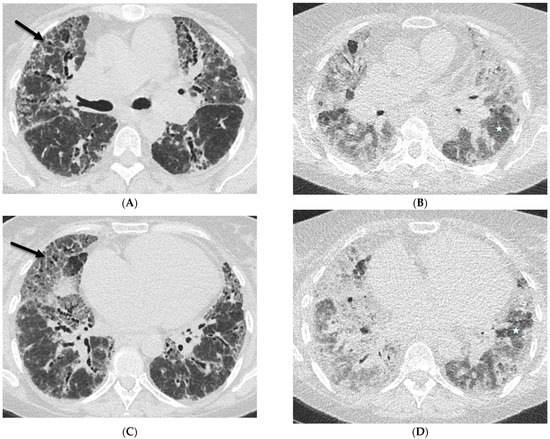
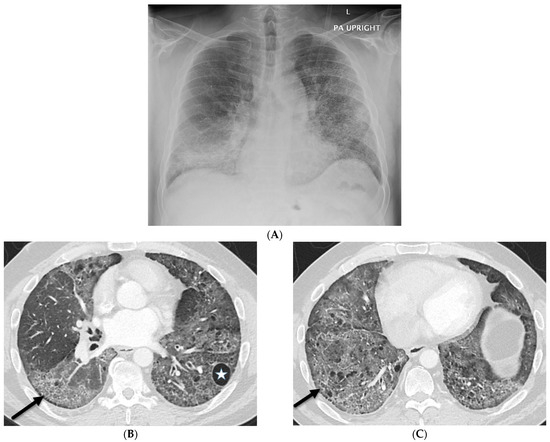
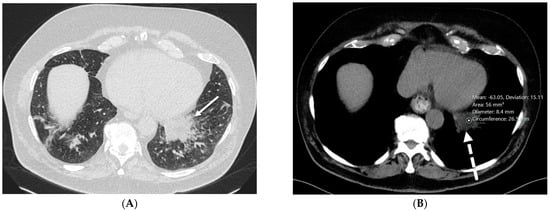
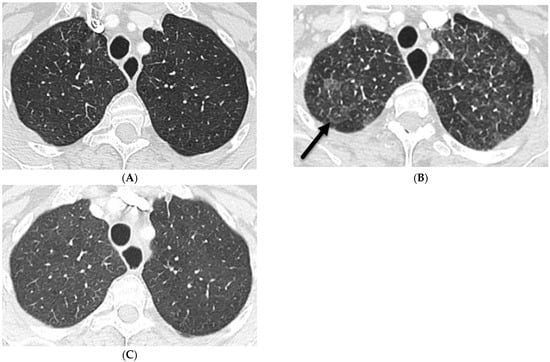
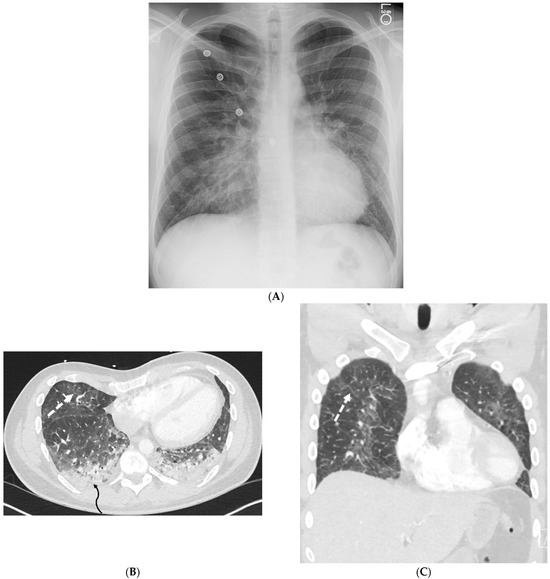
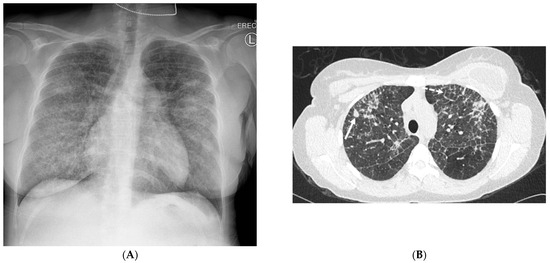
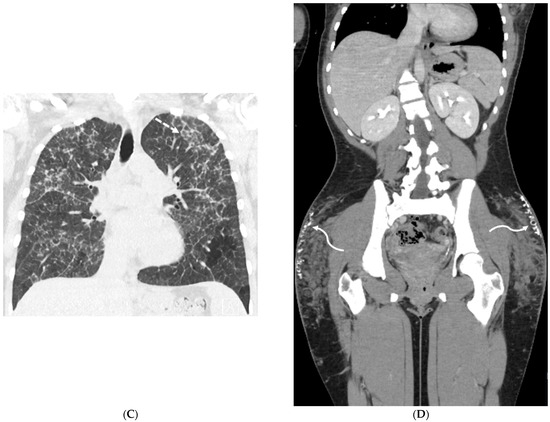
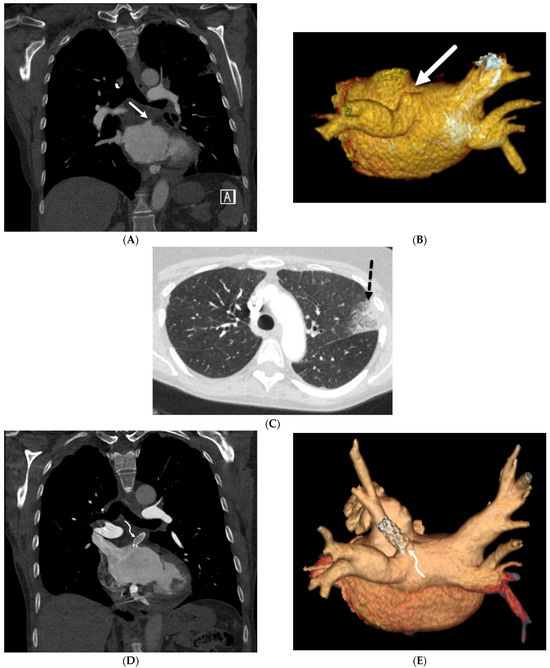
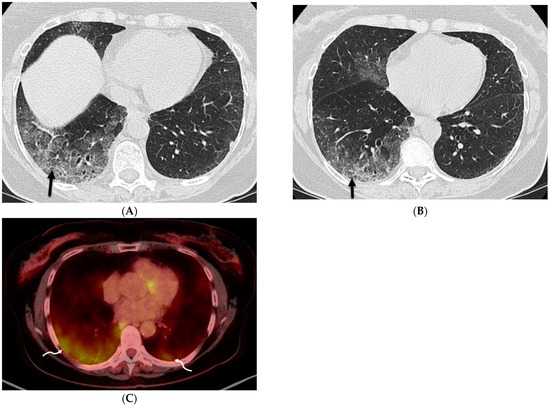
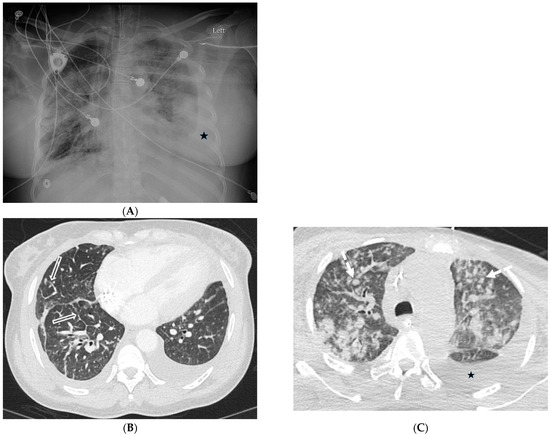
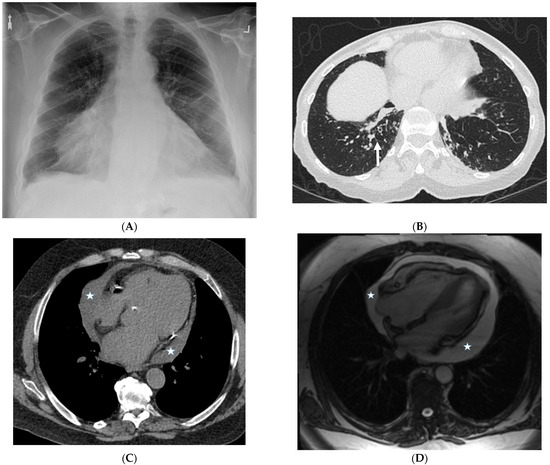
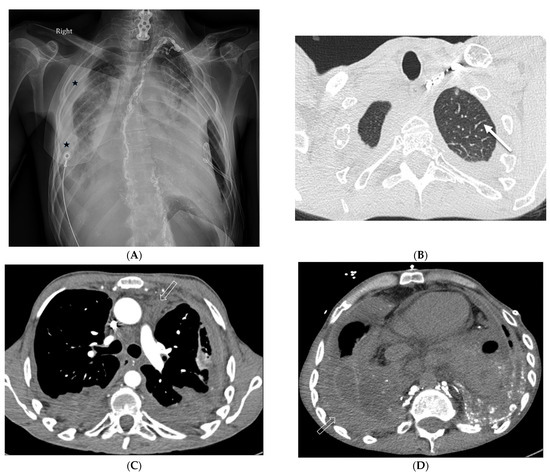
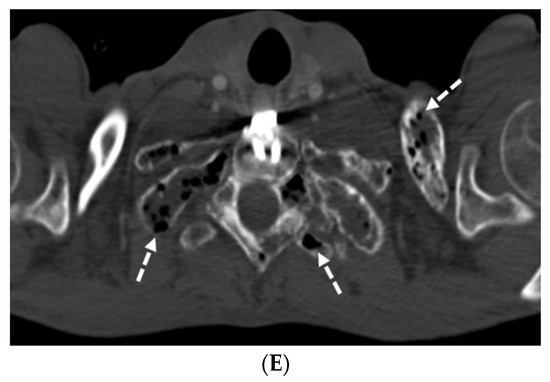
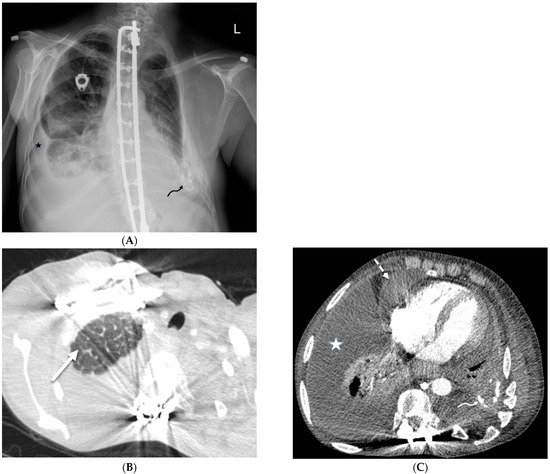
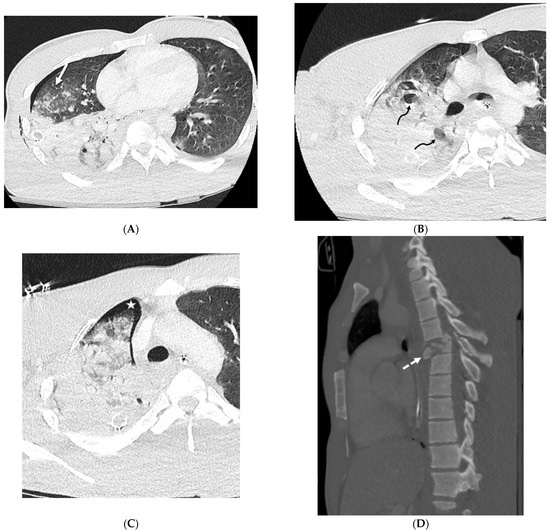
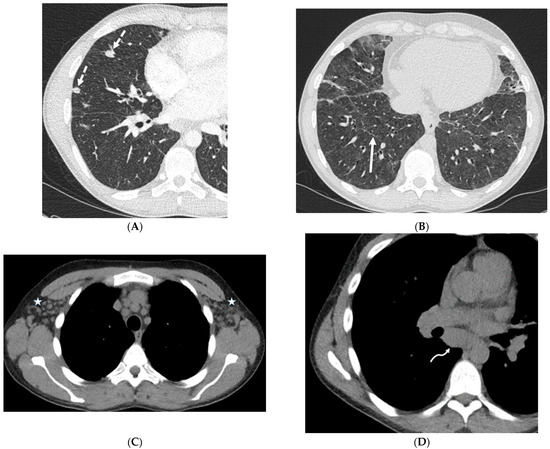
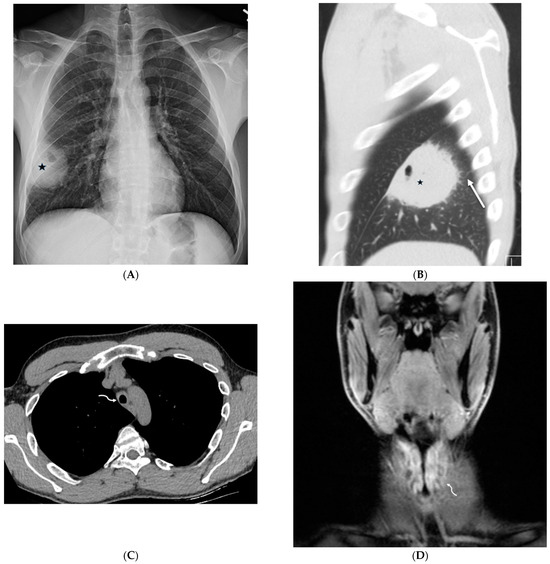
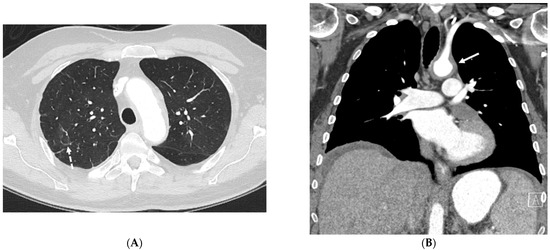
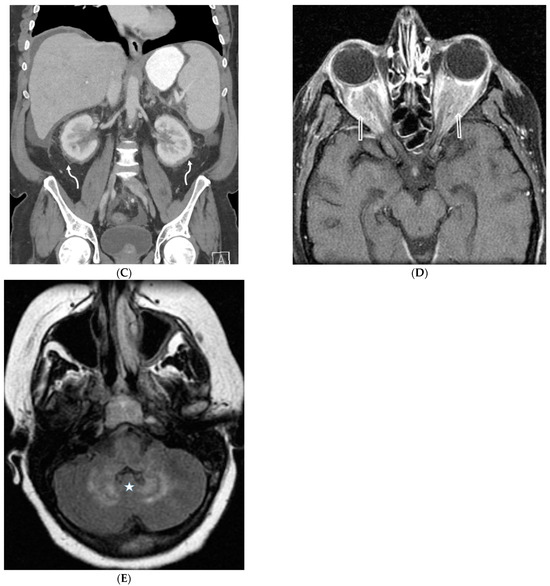
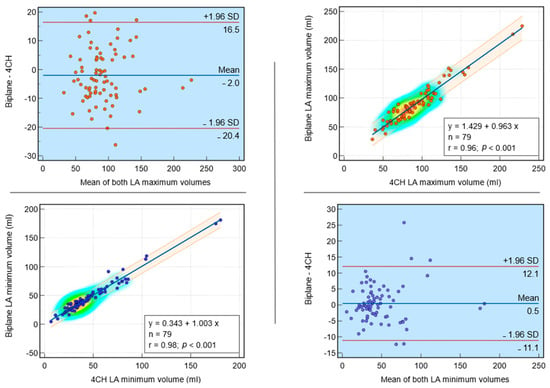
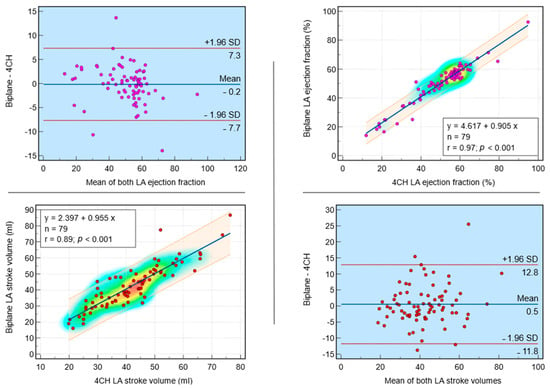
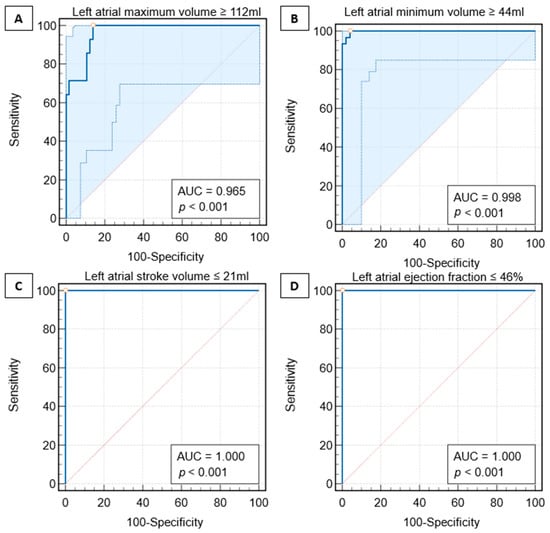
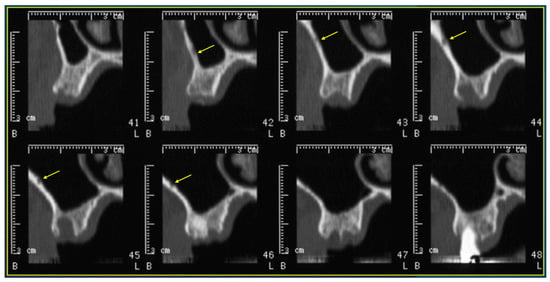
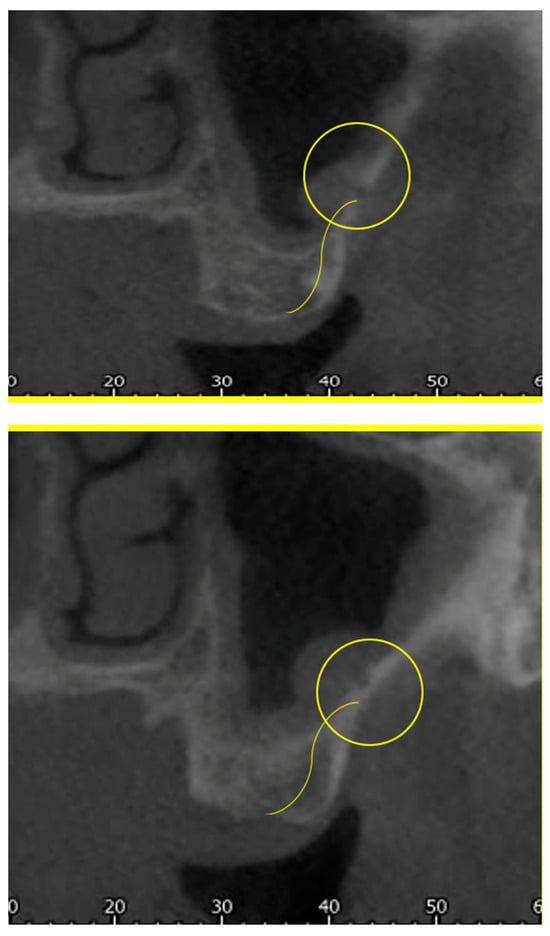
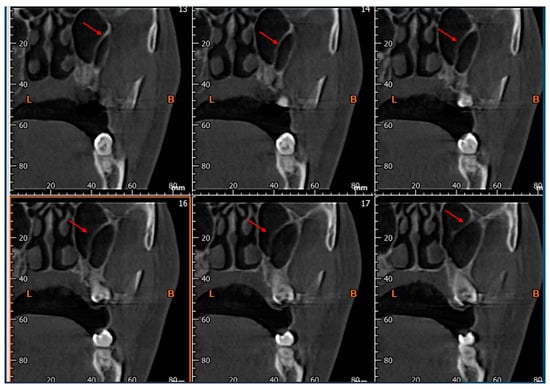
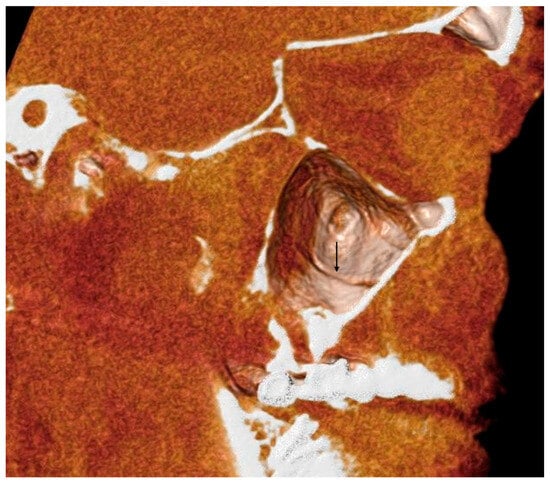
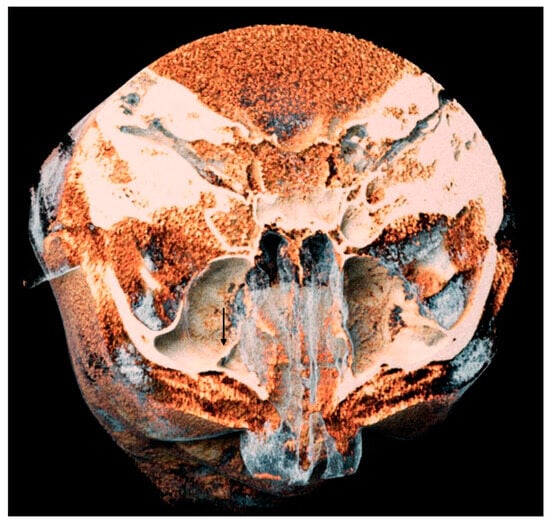
Interlobular septa thickening (ILST) is a common and easily recognized feature on computed tomography (CT) images in many lung disorders. ILST thickening can be smooth (most common), nodular, or irregular. Smooth ILST can be seen in pulmonary edema, pulmonary alveolar proteinosis, and lymphangitic spread of tumors. Nodular ILST can be seen in the lymphangitic spread of tumors, sarcoidosis, and silicosis. Irregular ILST is a finding suggestive of interstitial fibrosis, which is a common finding in fibrotic lung diseases, including sarcoidosis and usual interstitial pneumonia. Pulmonary edema and lymphangitic spread of tumors are the commonly encountered causes of ILST. It is important to narrow down the differential diagnosis as much as possible by assessing the appearance and distribution of ILST, as well as other pulmonary and extrapulmonary findings. This review will focus on the CT characterization of the secondary pulmonary lobule and ILST. Various uncommon causes of ILST will be discussed, including infections, interstitial pneumonia, depositional/infiltrative conditions, inhalational disorders, malignancies, congenital/inherited conditions, and iatrogenic causes. Awareness of the imaging appearance and various causes of ILST allows for a systematic approach, which is important for a timely diagnosis. This study highlights the importance of a structured approach to CT scan analysis that considers ILST characteristics, associated findings, and differential diagnostic considerations to facilitate accurate diagnoses.
Full article
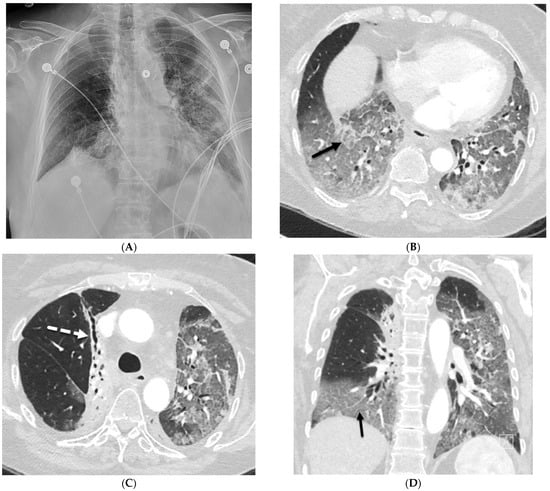
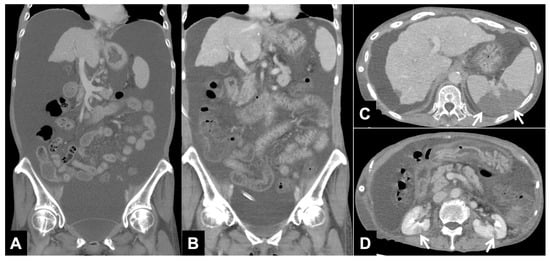
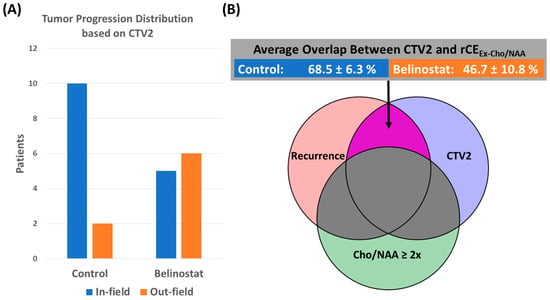
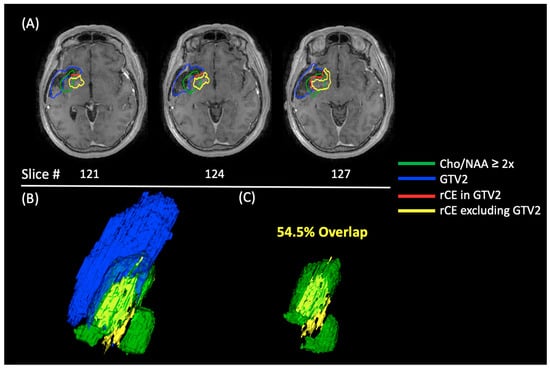
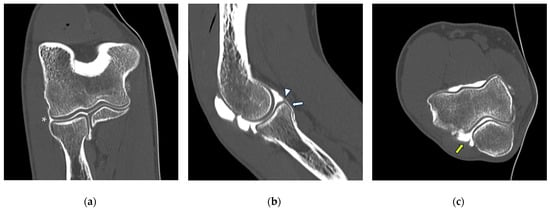
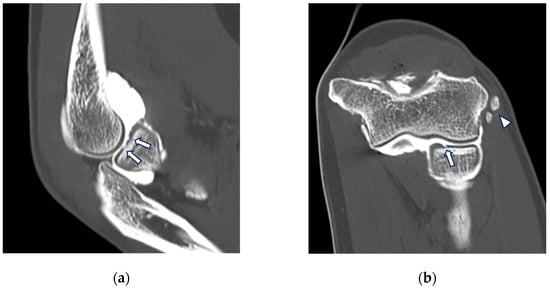
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}