


Tomography 2024, 10(4), 632-642; https://doi.org/10.3390/tomography10040048 - 22 Apr 2024
Abstract
Rationale: F18-FDG PET/CT may be helpful in baseline staging of patients with high-risk LARC presenting with vascular tumor deposits (TDs), in addition to standard pelvic MRI and CT staging. Methods: All patients with locally advanced rectal cancer that had TDs on their baseline
[...] Read more.
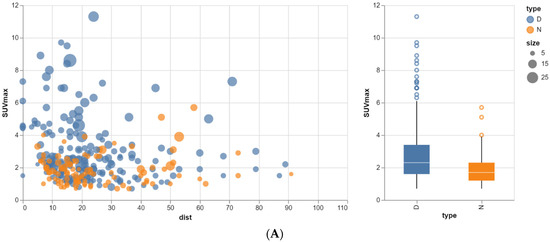
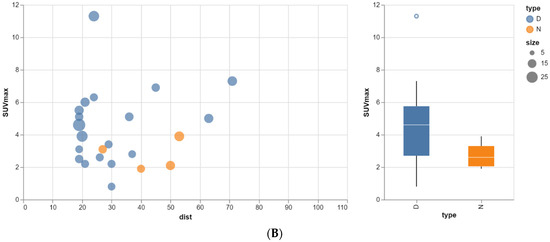
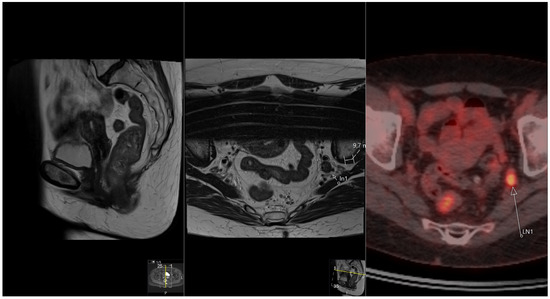
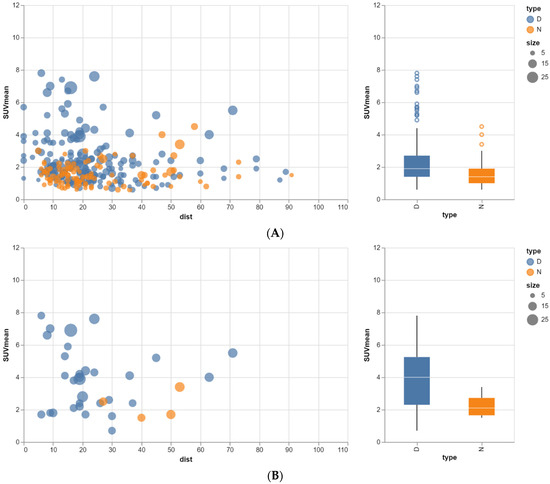
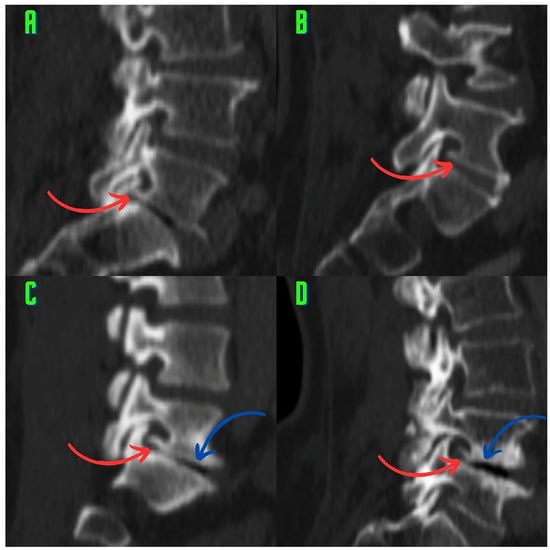
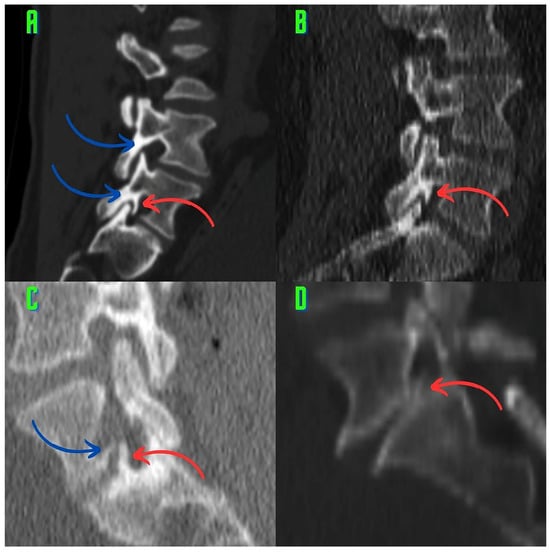
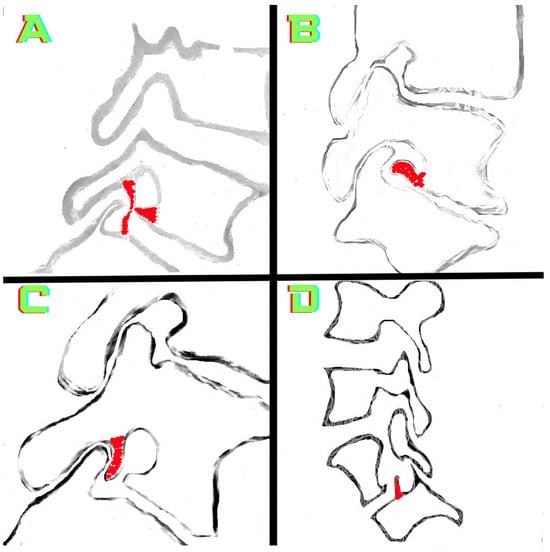
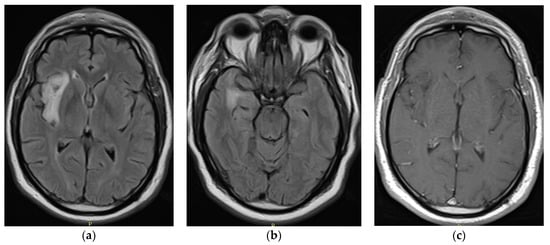
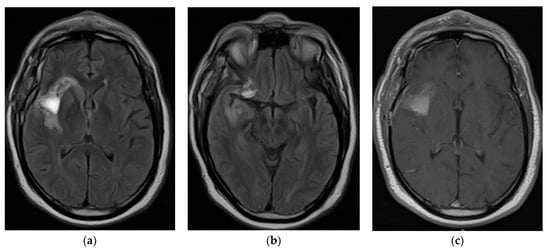
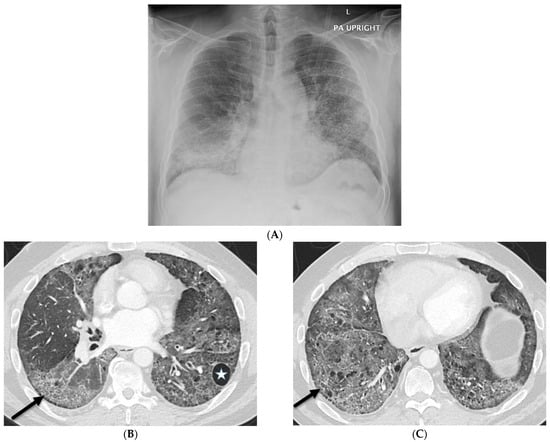
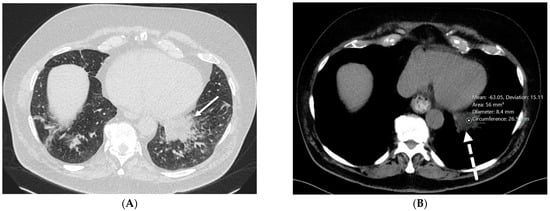
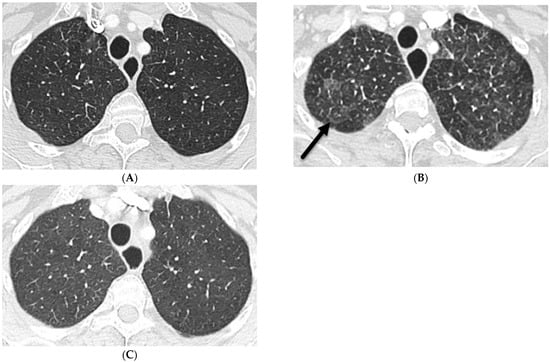
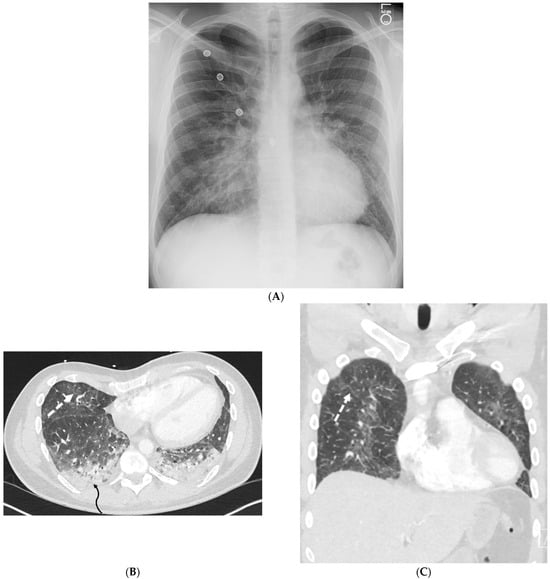
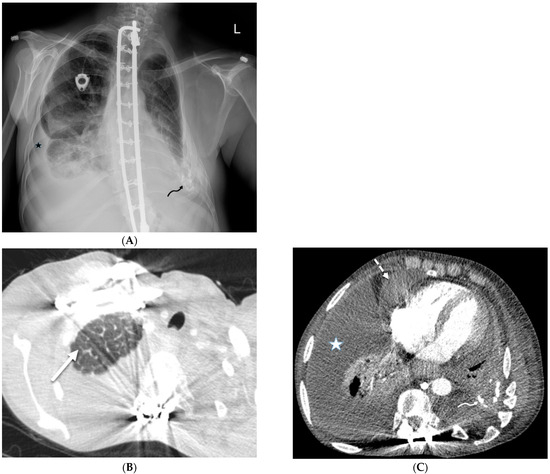
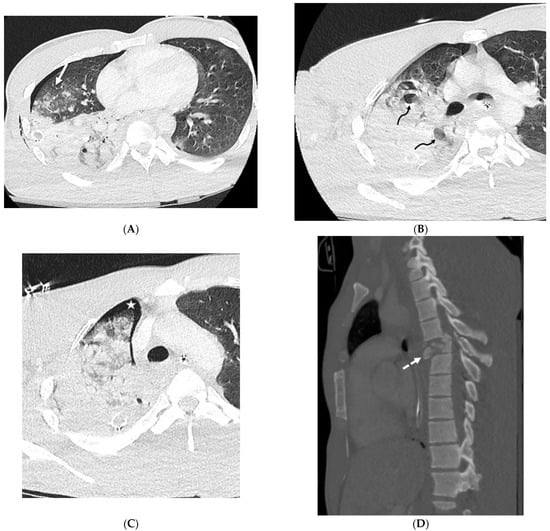
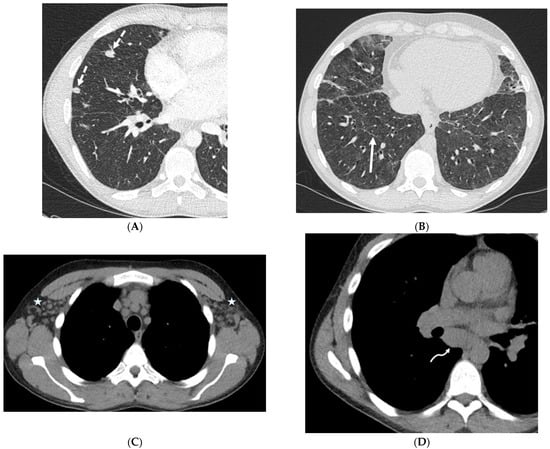
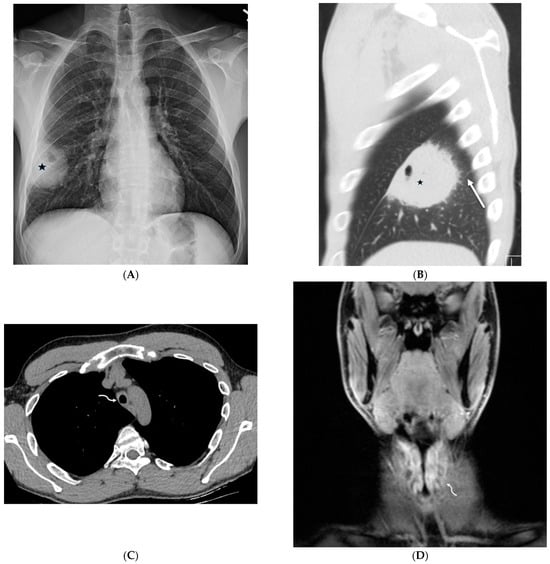
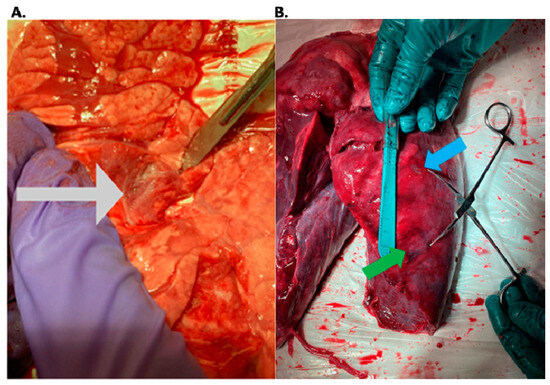
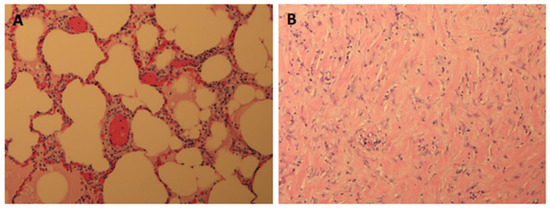
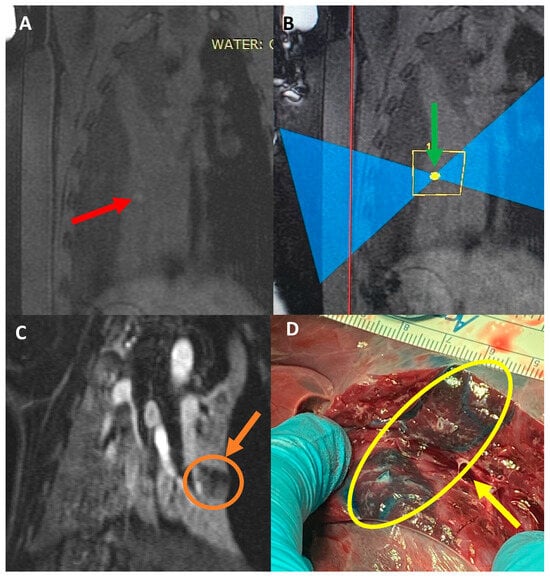
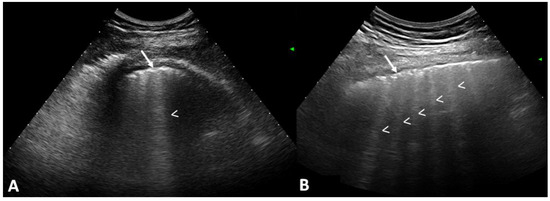
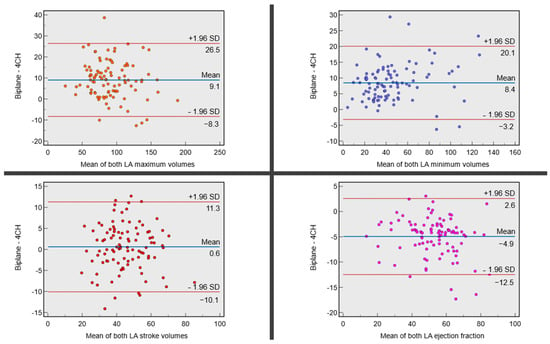
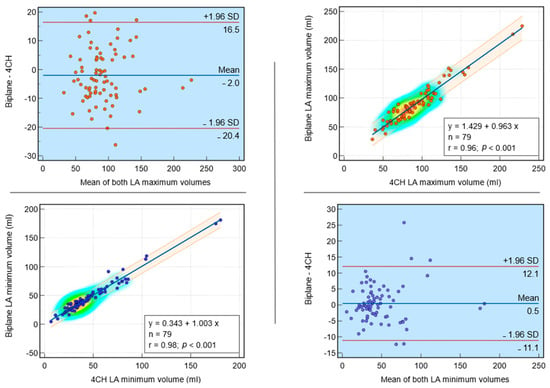
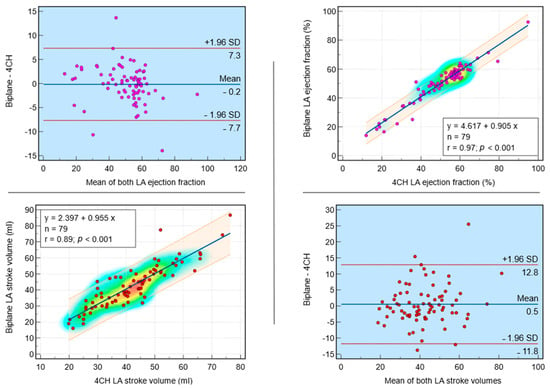
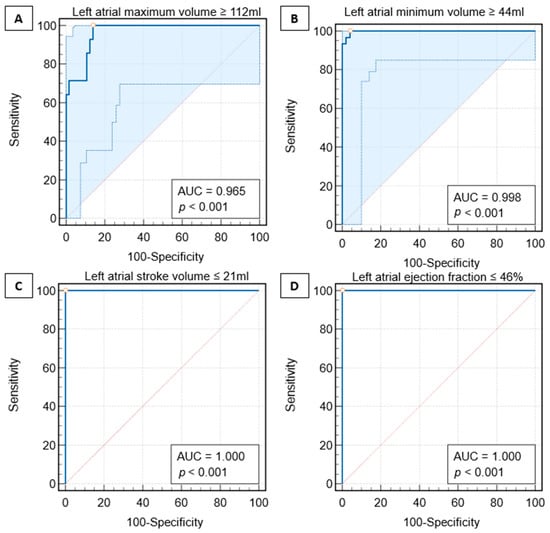
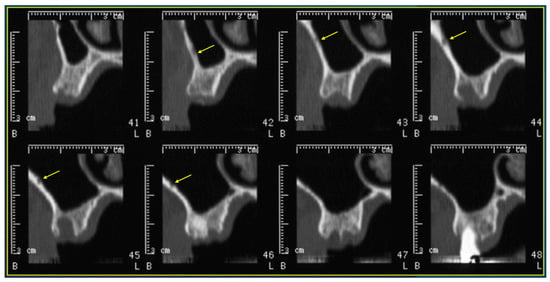
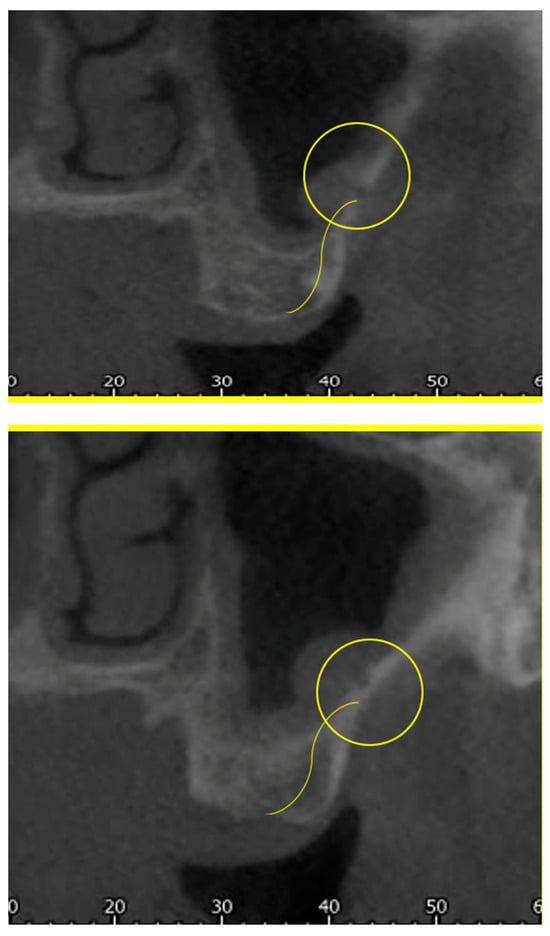
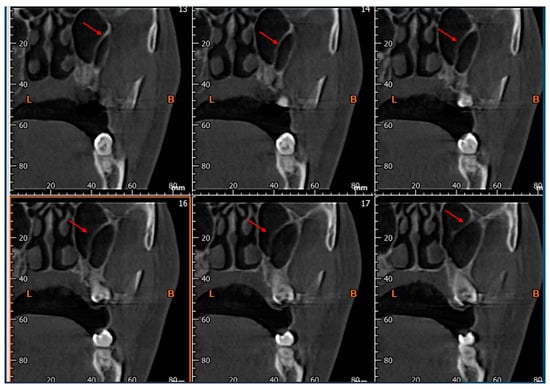
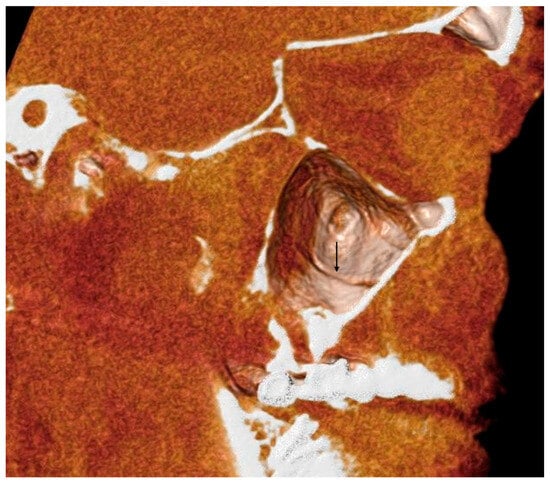
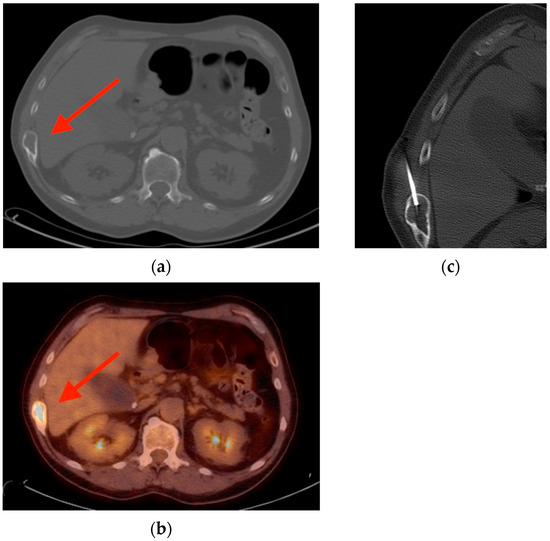
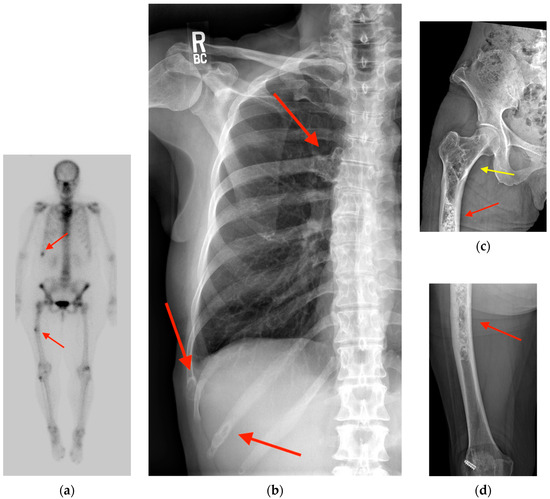
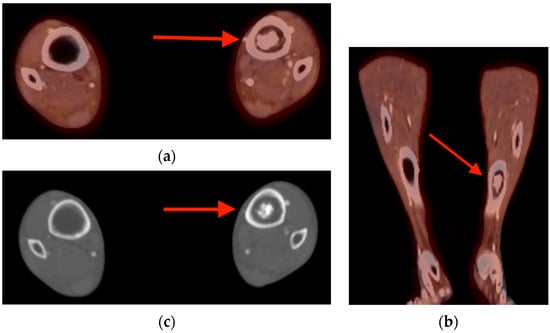
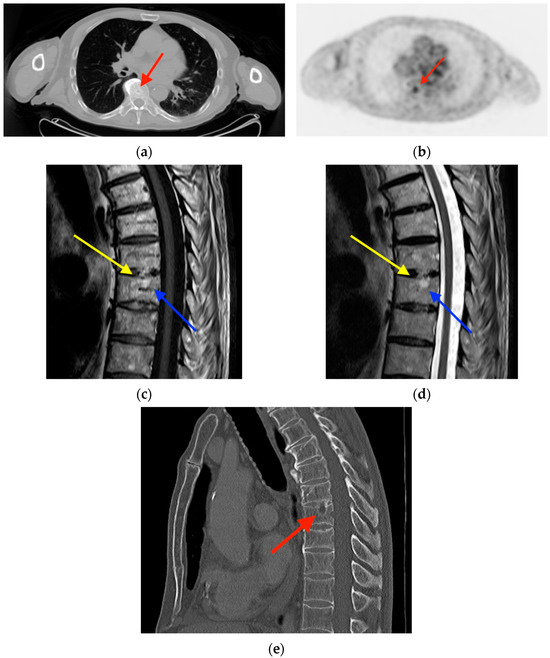
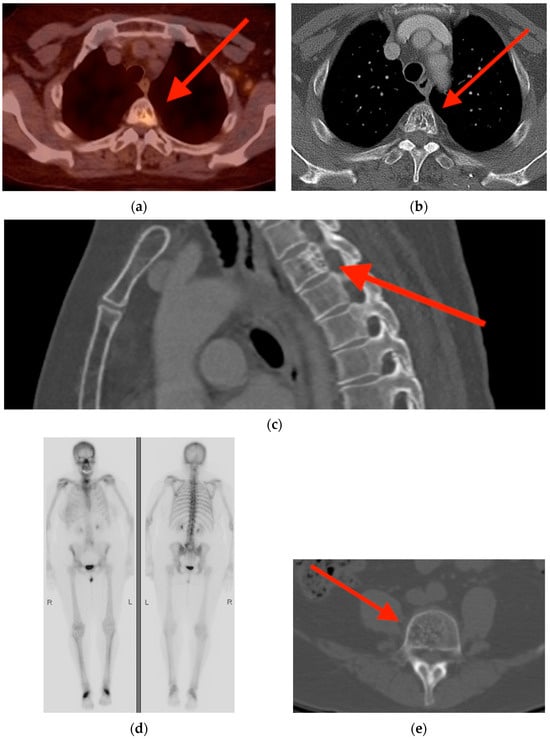
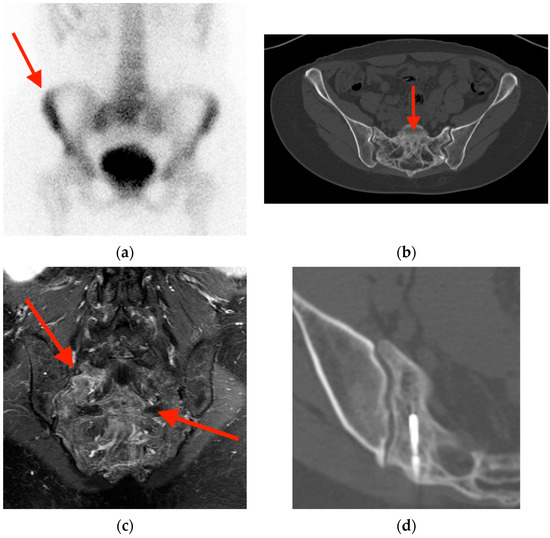
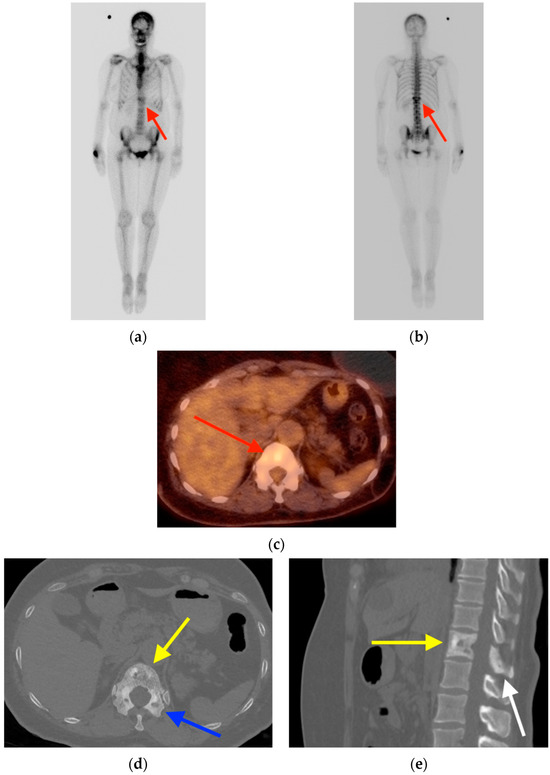
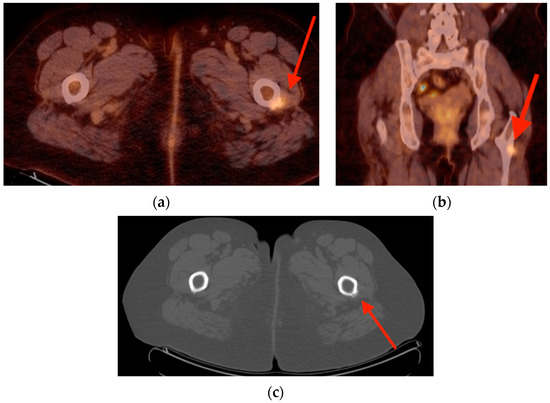
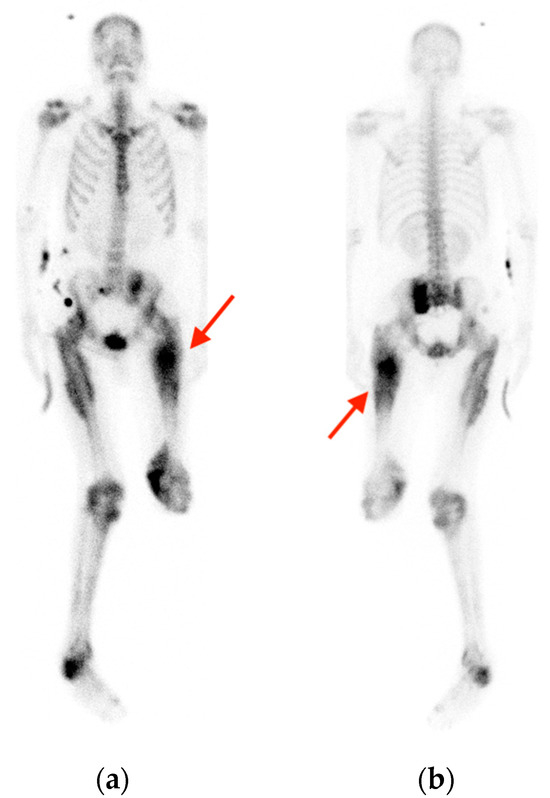
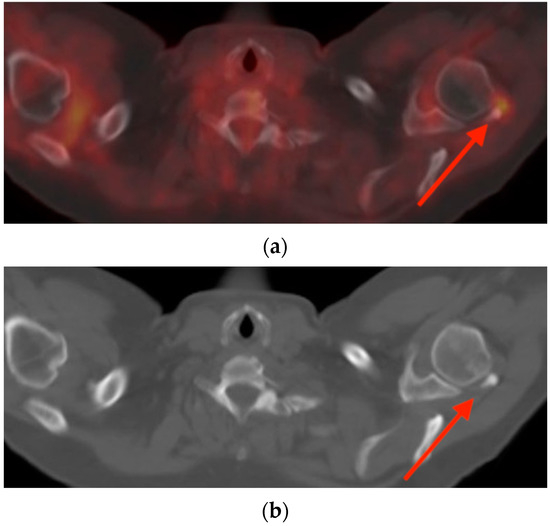
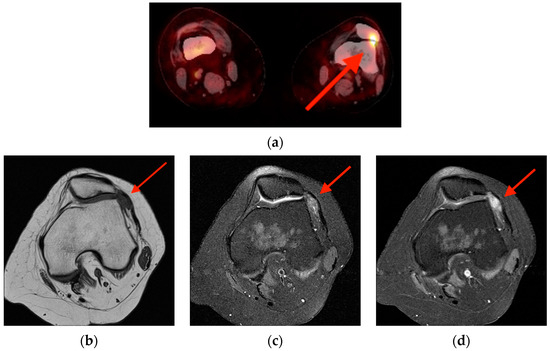
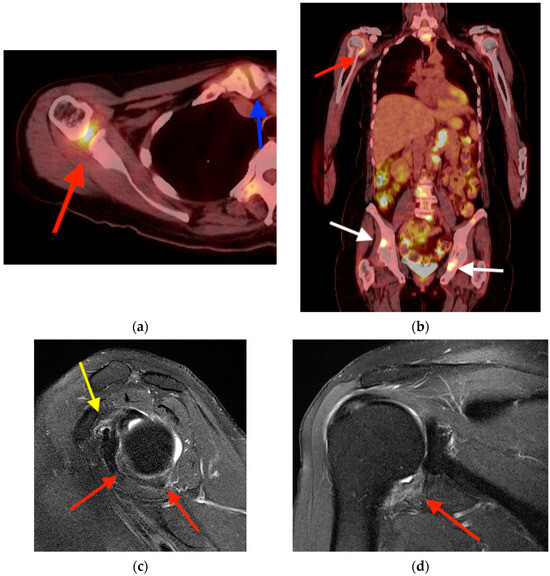
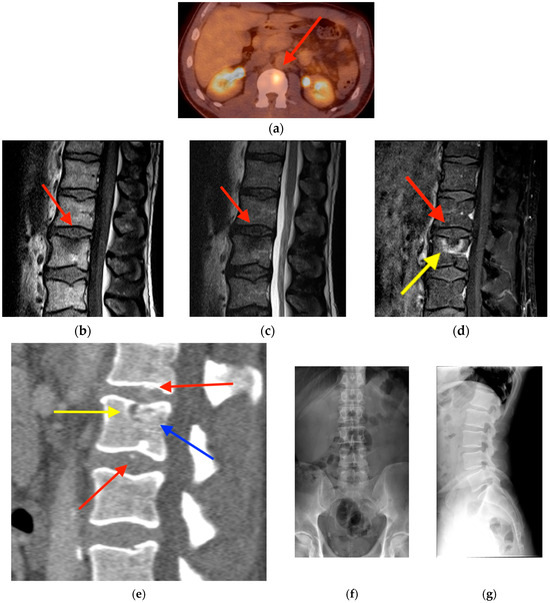
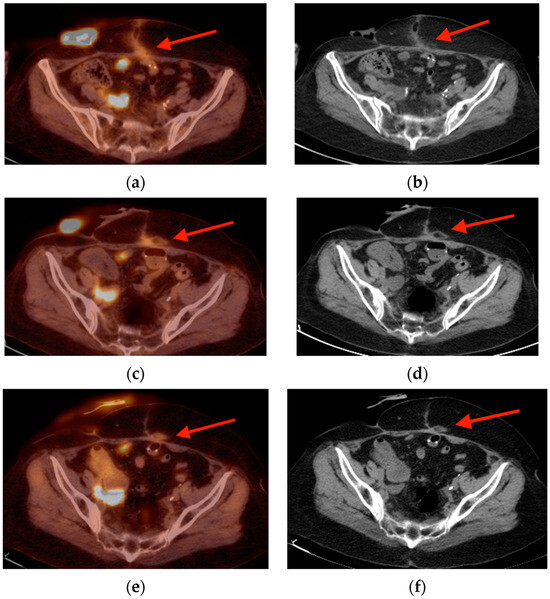
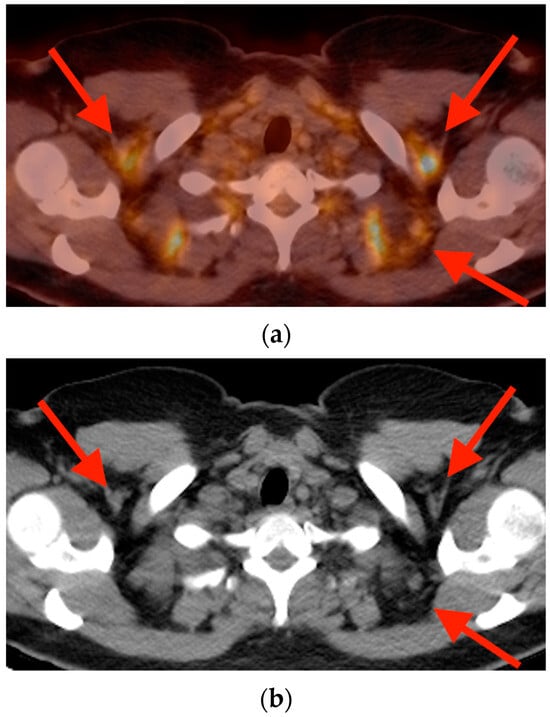
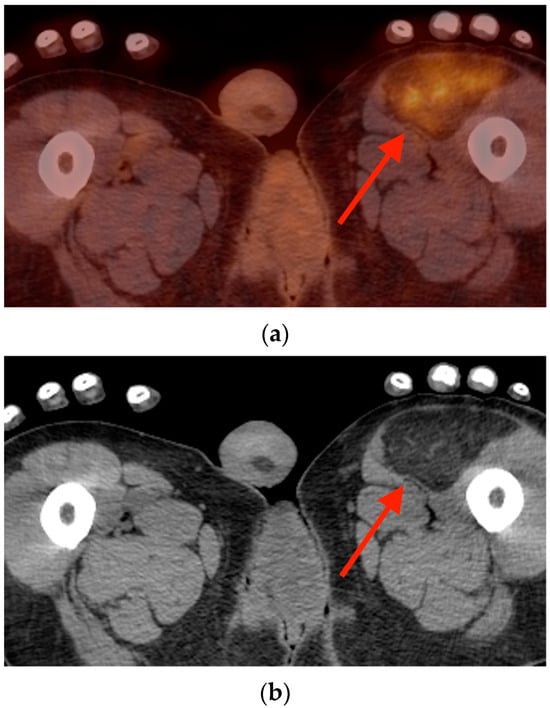
Rationale: F18-FDG PET/CT may be helpful in baseline staging of patients with high-risk LARC presenting with vascular tumor deposits (TDs), in addition to standard pelvic MRI and CT staging. Methods: All patients with locally advanced rectal cancer that had TDs on their baseline MRI of the pelvis and had a baseline F18-FDG PET/CT between May 2016 and December 2020 were included in this retrospective study. TDs as well as lymph nodes identified on pelvic MRI were correlated to the corresponding nodular structures on a standard F18-FDG PET/CT, including measurements of nodular SUVmax and SUVmean. In addition, the effects of partial volume and spill-in on SUV measurements were studied. Results: A total number of 62 patients were included, in which 198 TDs were identified as well as 106 lymph nodes (both normal and metastatic). After ruling out partial volume effects and spill-in, 23 nodular structures remained that allowed for reliable measurement of SUVmax: 19 TDs and 4 LNs. The median SUVmax between TDs and LNs was not significantly different (p = 0.096): 4.6 (range 0.8 to 11.3) versus 2.8 (range 1.9 to 3.9). For the median SUVmean, there was a trend towards a significant difference (p = 0.08): 3.9 (range 0.7 to 7.8) versus 2.3 (range 1.5 to 3.4). Most nodular structures showing either an SUVmax or SUVmean ≥ 4 were characterized as TDs on MRI, while only two were characterized as LNs. Conclusions: SUV measurements may help in separating TDs from lymph node metastases or normal lymph nodes in patients with high-risk LARC.
Full article
(This article belongs to the Special Issue Functional and Molecular Imaging of the Abdomen)
►
Show Figures

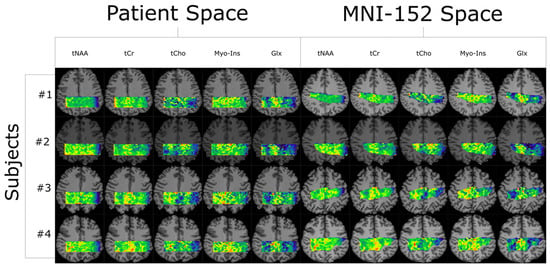
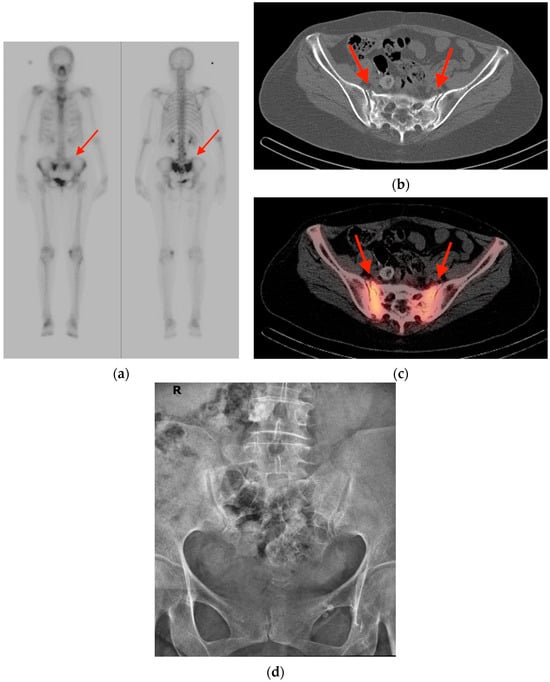
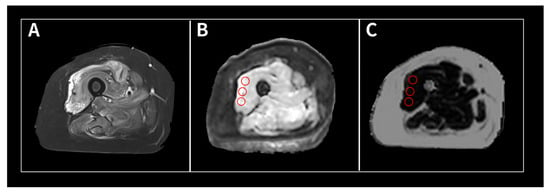
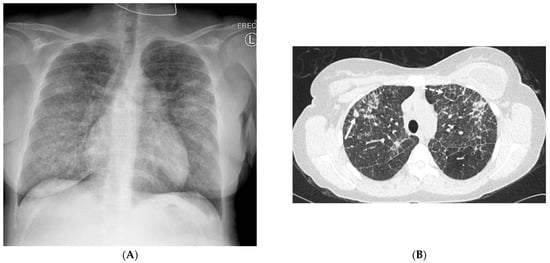
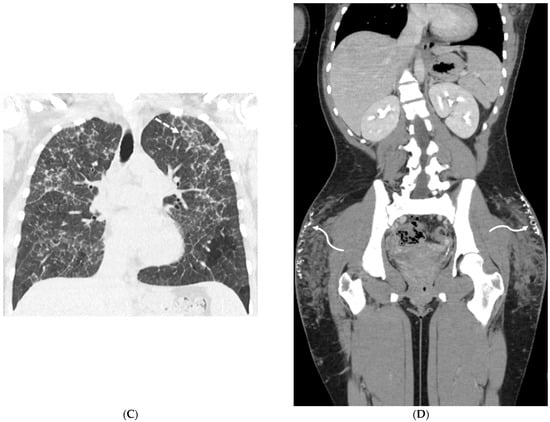
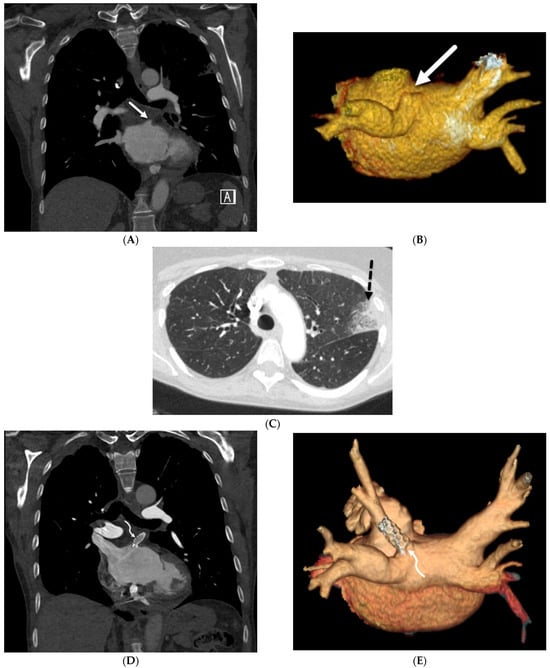
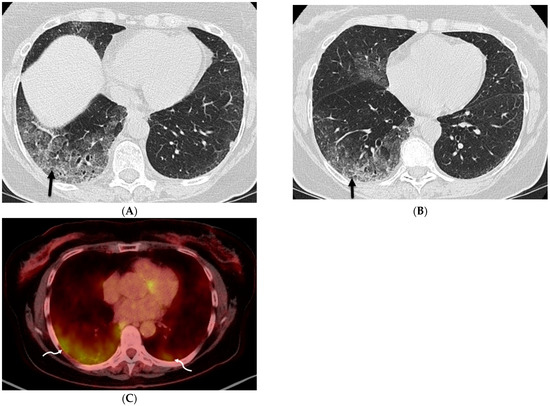
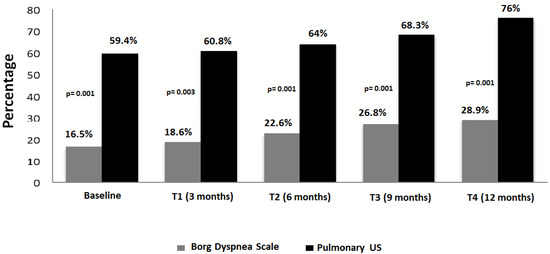
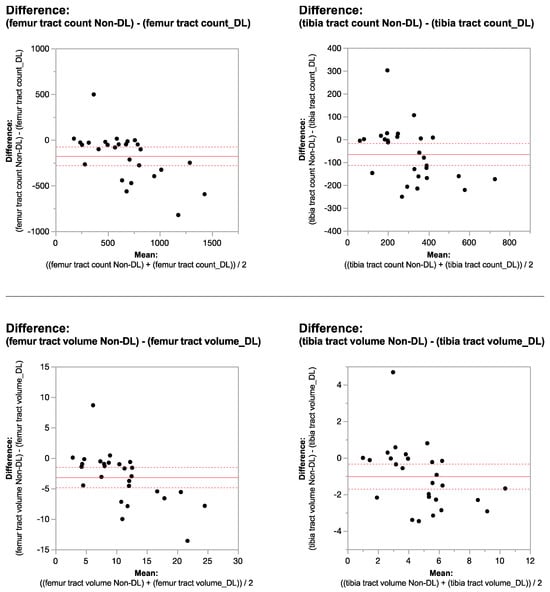
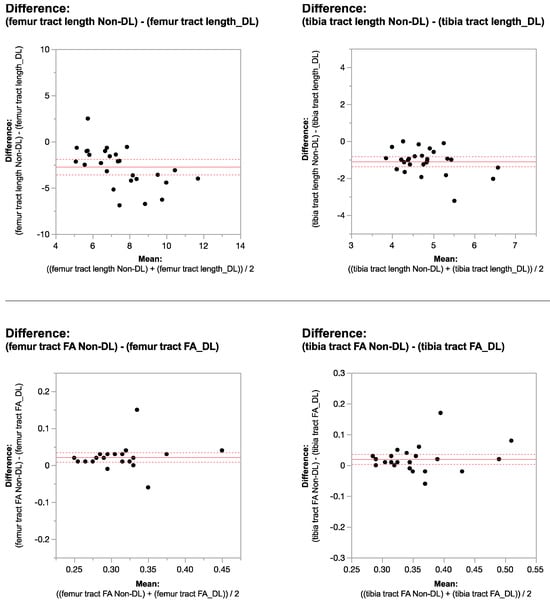
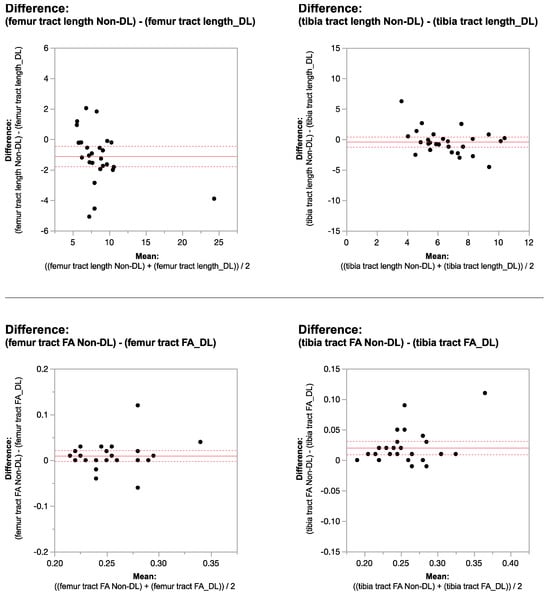
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}