


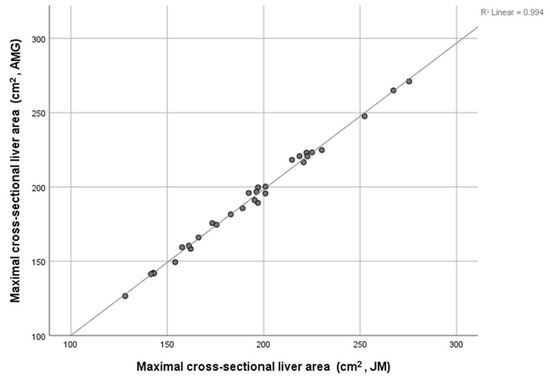
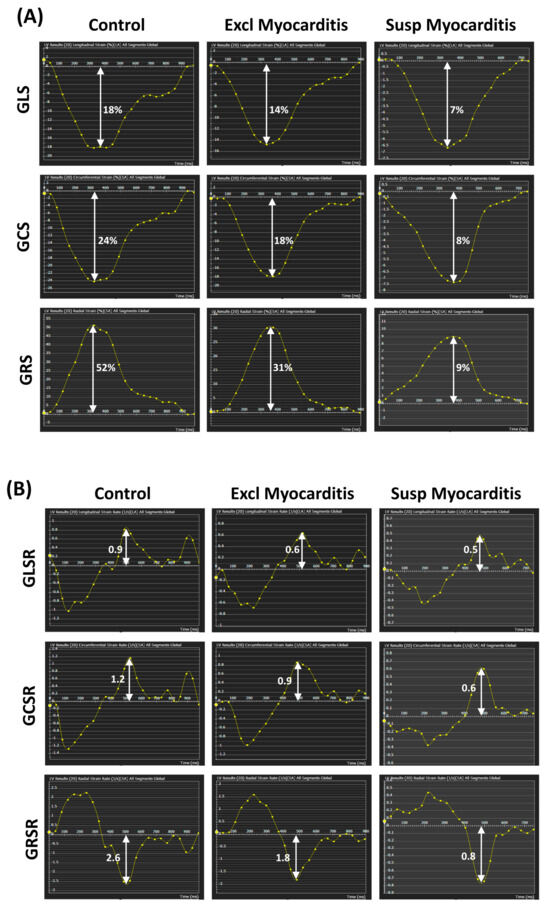
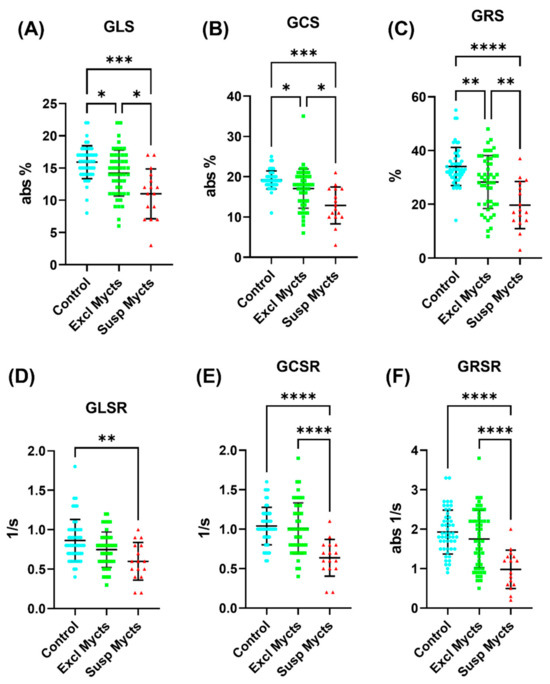
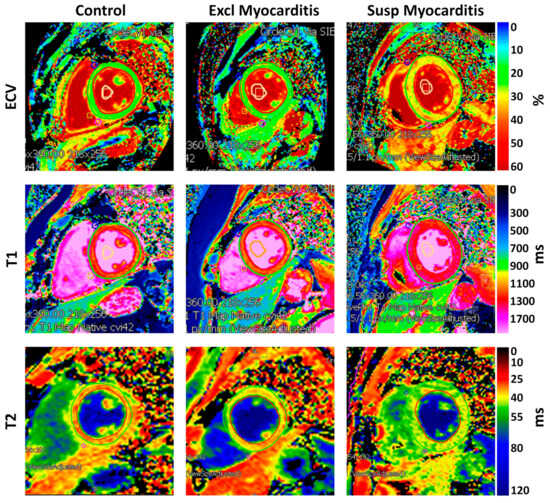
Tomography 2024, 10(4), 554-573; https://doi.org/10.3390/tomography10040044 - 15 Apr 2024
Abstract
►
Show Figures
This review provides unique insights to the scientific scope and clinical visions of the inventors and pioneers of the SoftVue breast tomographic ultrasound (BTUS). Their >20-year collaboration produced extensive basic research and technology developments, culminating in SoftVue, which recently received the Food and
[...] Read more.
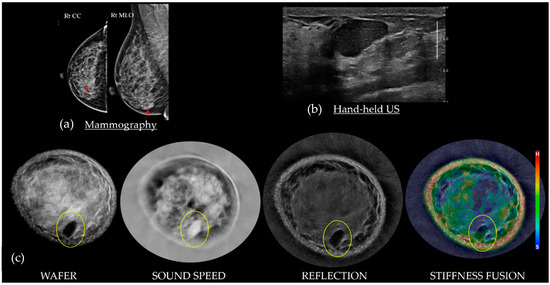
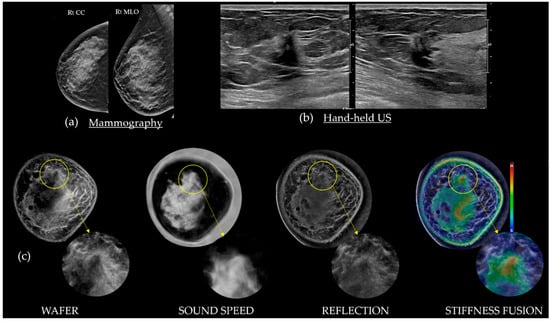
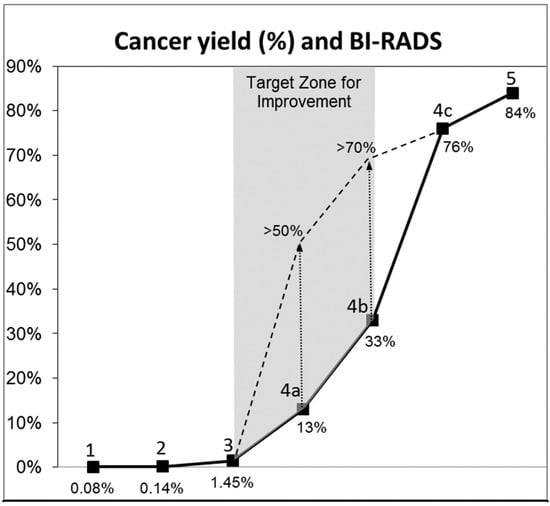
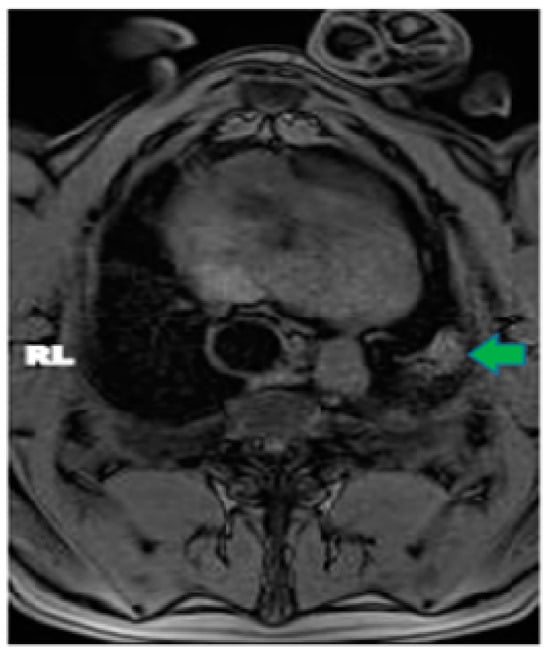
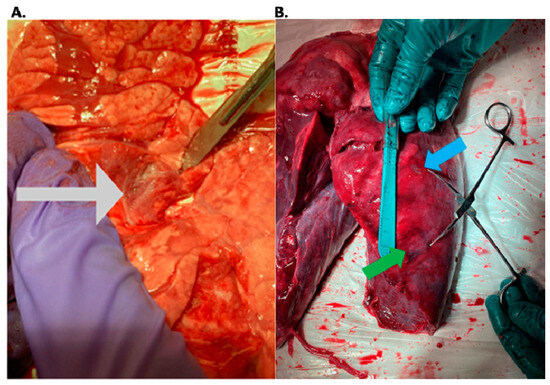
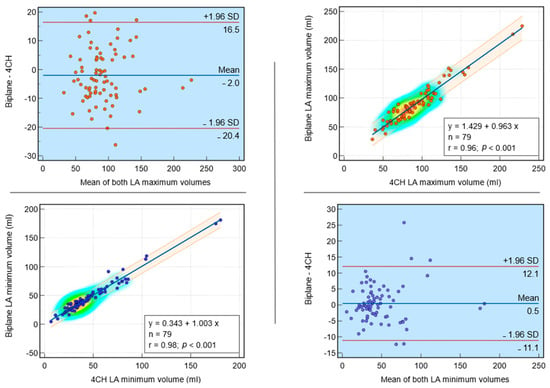
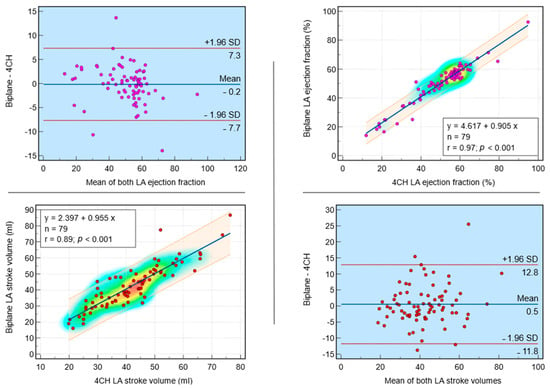
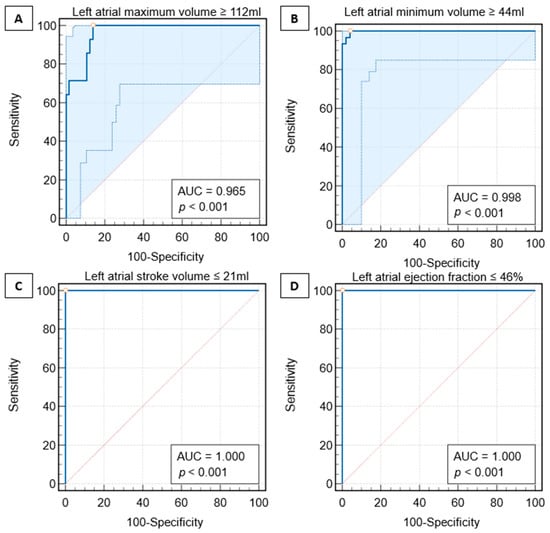
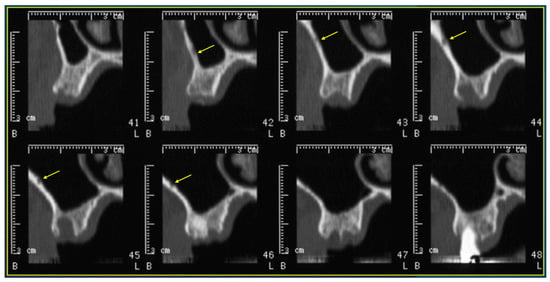
This review provides unique insights to the scientific scope and clinical visions of the inventors and pioneers of the SoftVue breast tomographic ultrasound (BTUS). Their >20-year collaboration produced extensive basic research and technology developments, culminating in SoftVue, which recently received the Food and Drug Administration’s approval as an adjunct to breast cancer screening in women with dense breasts. SoftVue’s multi-center trial confirmed the diagnostic goals of the tissue characterization and localization of quantitative acoustic tissue differences in 2D and 3D coronal image sequences. SoftVue mass characterizations are also reviewed within the standard cancer risk categories of the Breast Imaging Reporting and Data System. As a quantitative diagnostic modality, SoftVue can also function as a cost-effective platform for artificial intelligence-assisted breast cancer identification. Finally, SoftVue’s quantitative acoustic maps facilitate noninvasive temperature monitoring and a unique form of time-reversed, focused US in a single theranostic device that actually focuses acoustic energy better within the highly scattering breast tissues, allowing for localized hyperthermia, drug delivery, and/or ablation. Women also prefer the comfort of SoftVue over mammograms and will continue to seek out less-invasive breast care, from diagnosis to treatment.
Full article
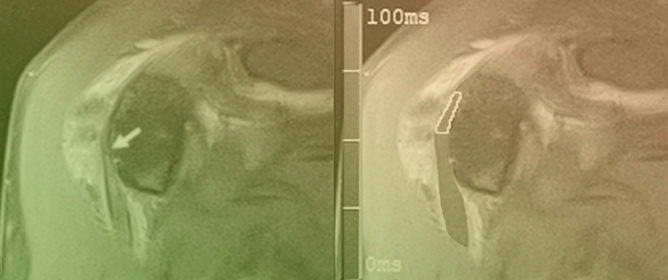
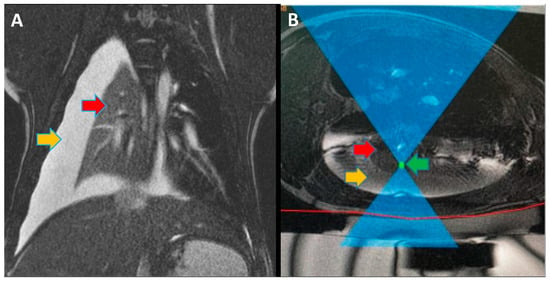
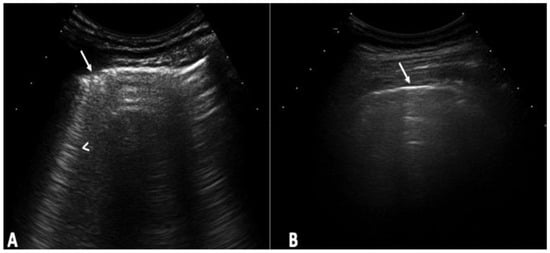
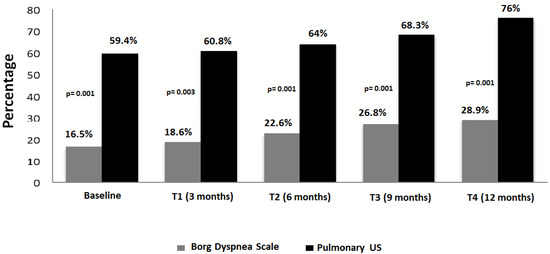
Figure 1




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
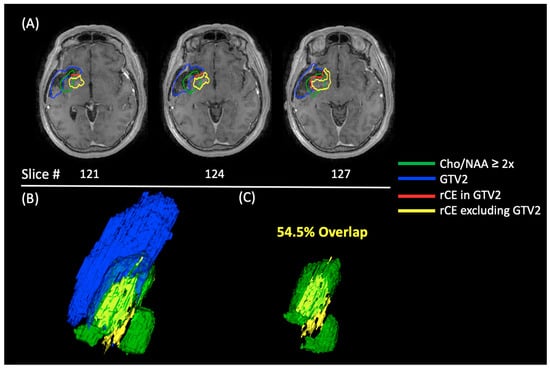
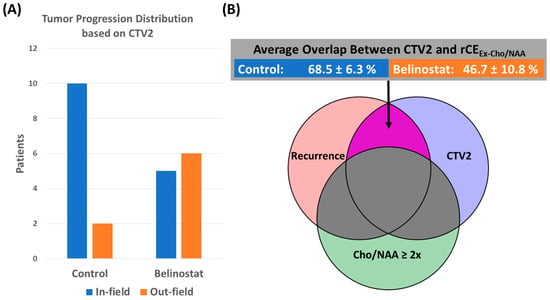{kind=link}
{kind=link}
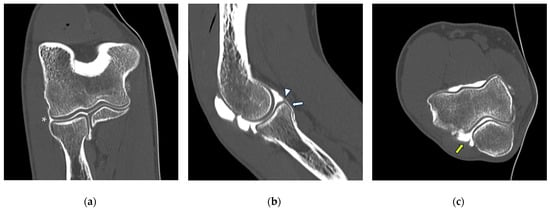
{kind=link}
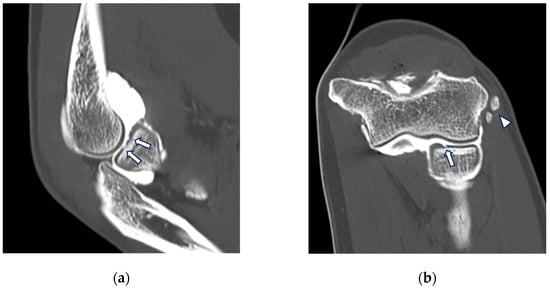
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}