


Tomography 2024, 10(5), 674-685; https://doi.org/10.3390/tomography10050052 - 01 May 2024
Abstract
The aim of this study was to evaluate the findings of CT scans in patients with pathologically confirmed primary colorectal squamous-cell carcinoma (SCC). The clinical presentation and CT findings in eight patients with pathologically confirmed primary colorectal squamous-cell carcinoma were retrospectively reviewed by
[...] Read more.
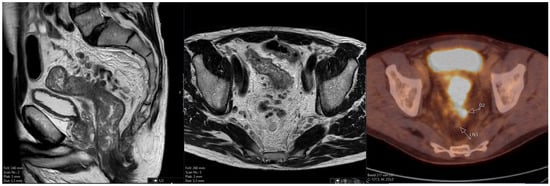
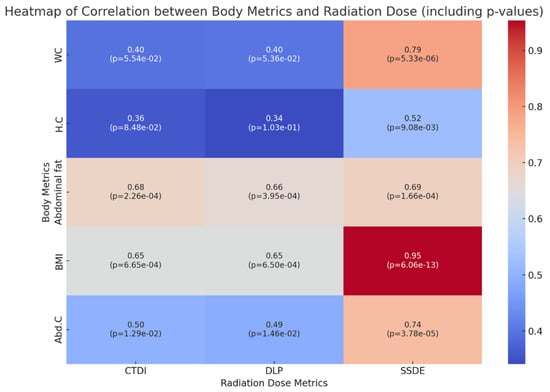
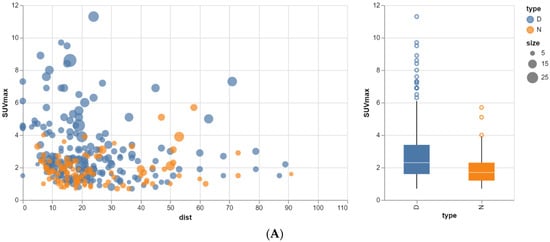
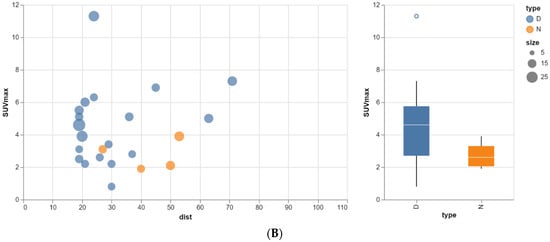
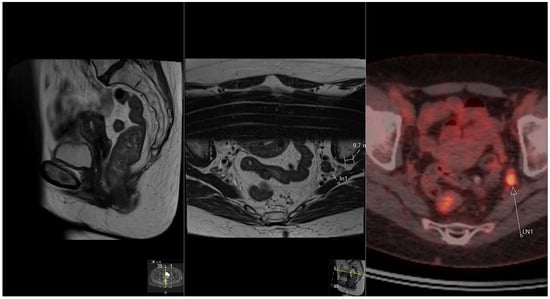
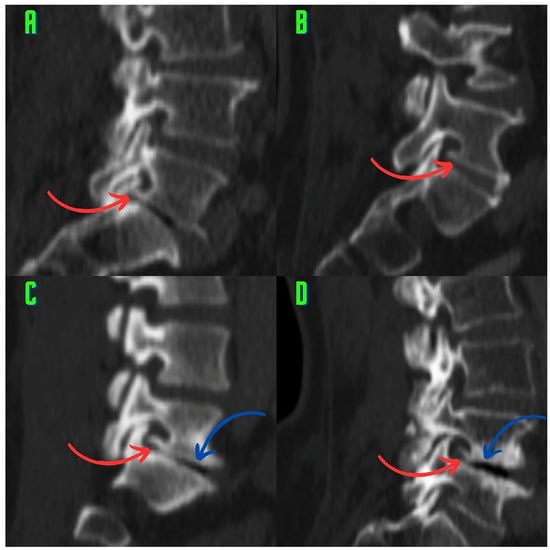
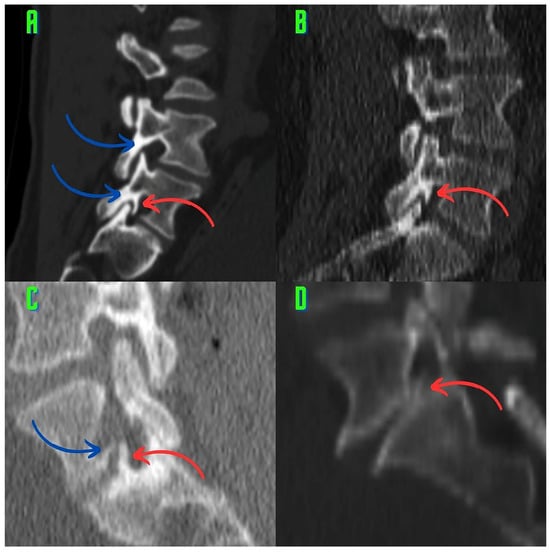
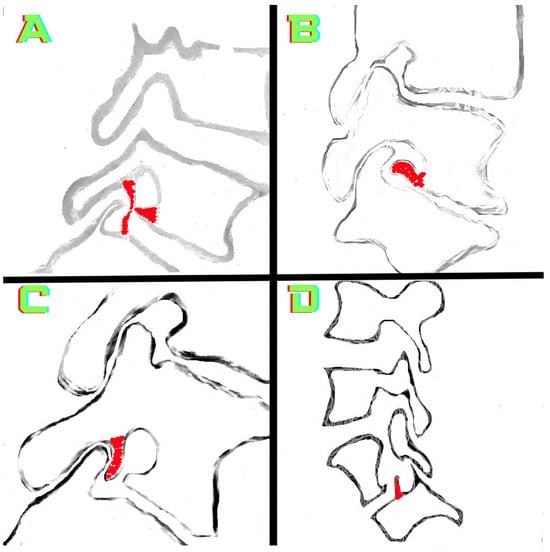
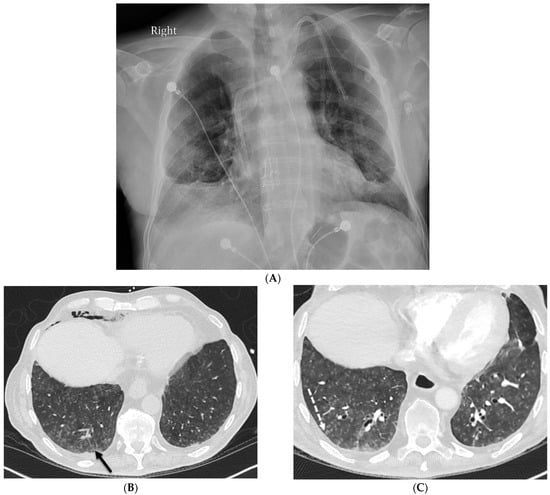
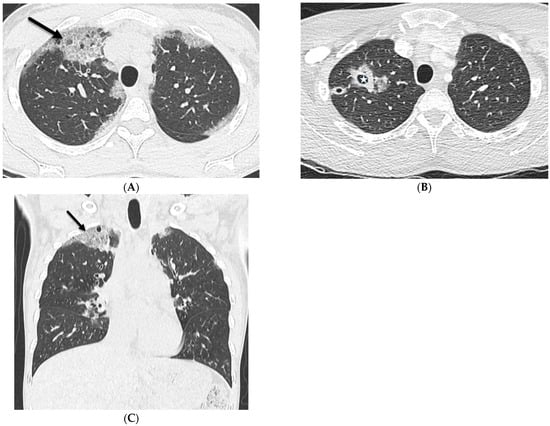
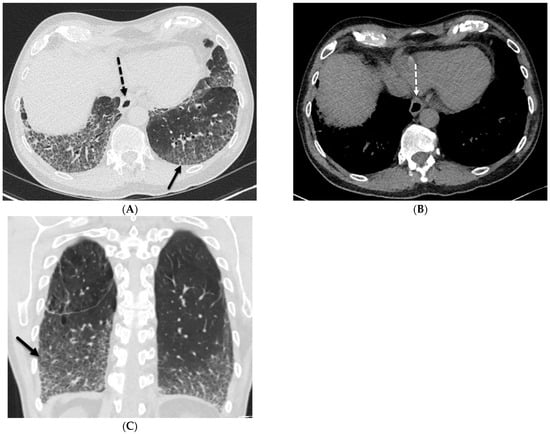
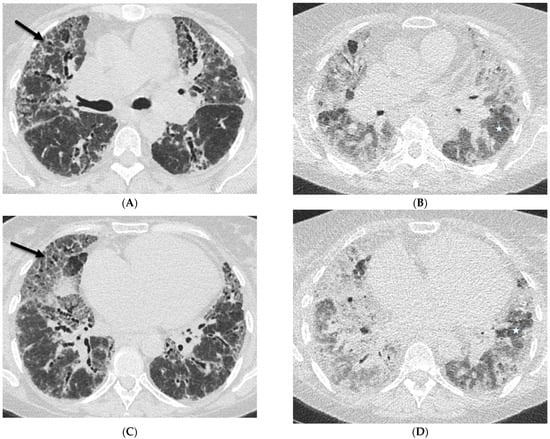
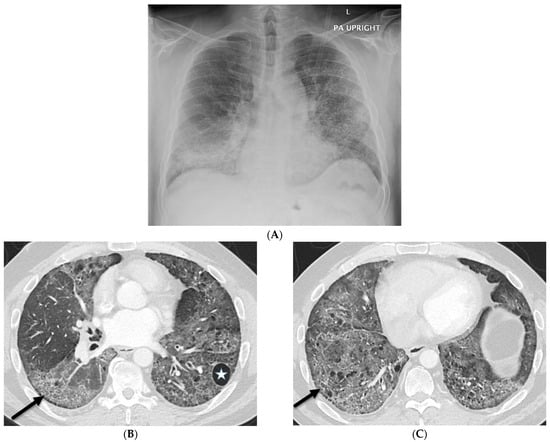
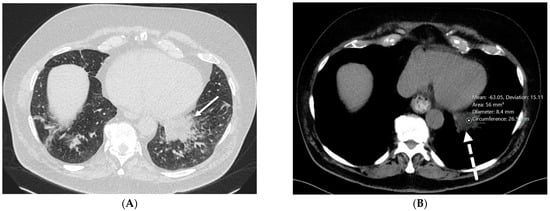
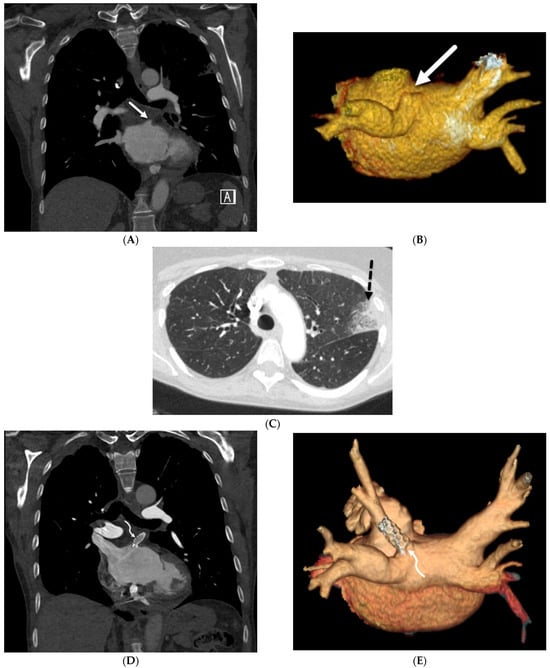
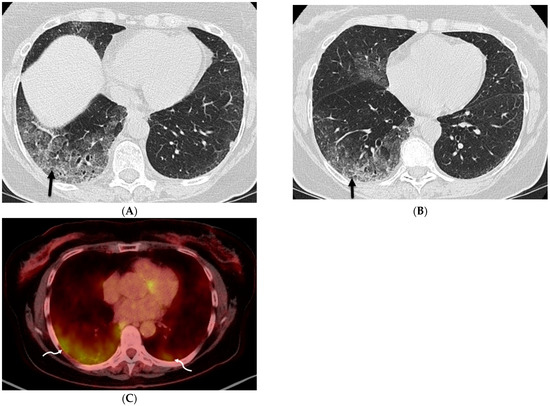
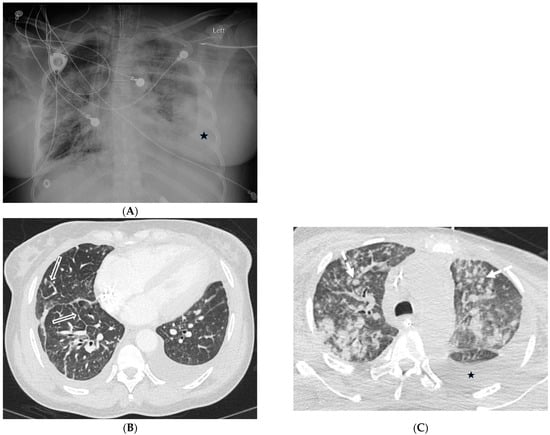
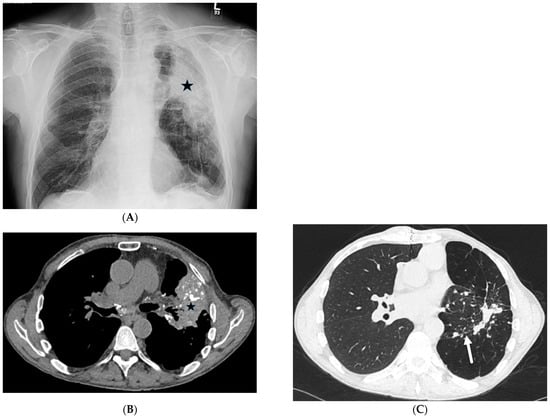
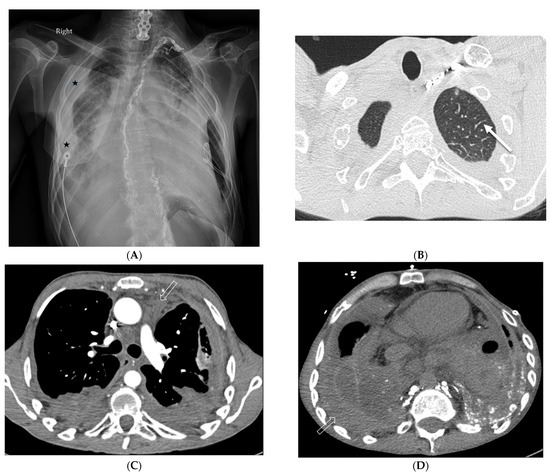
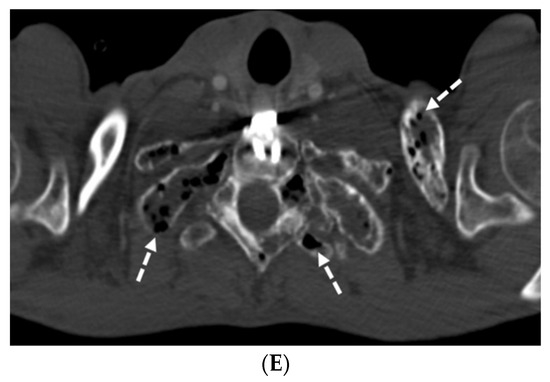
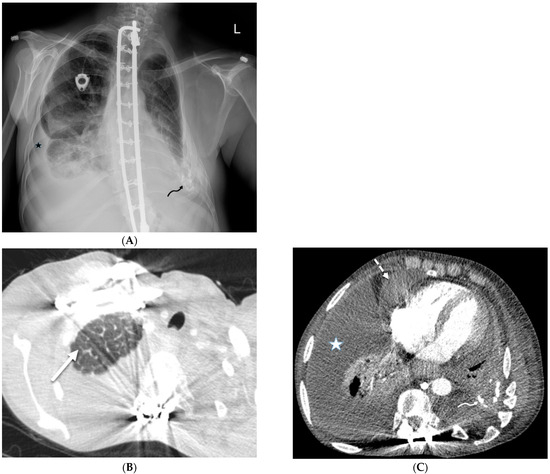
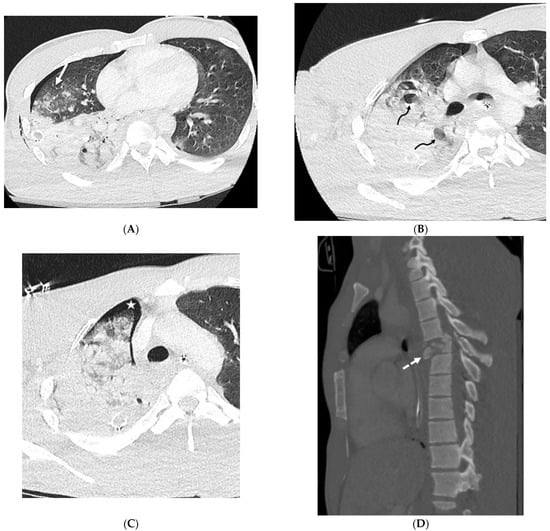
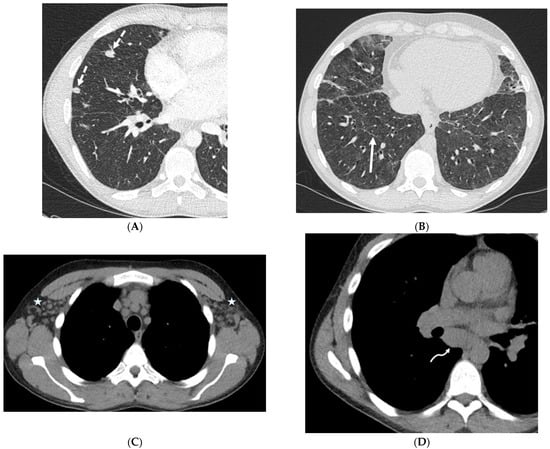
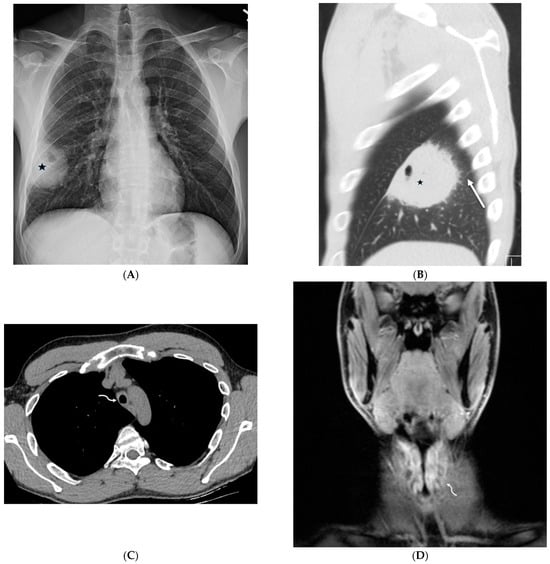
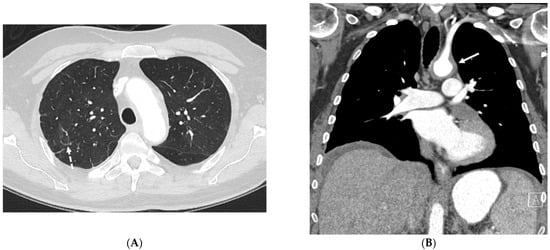
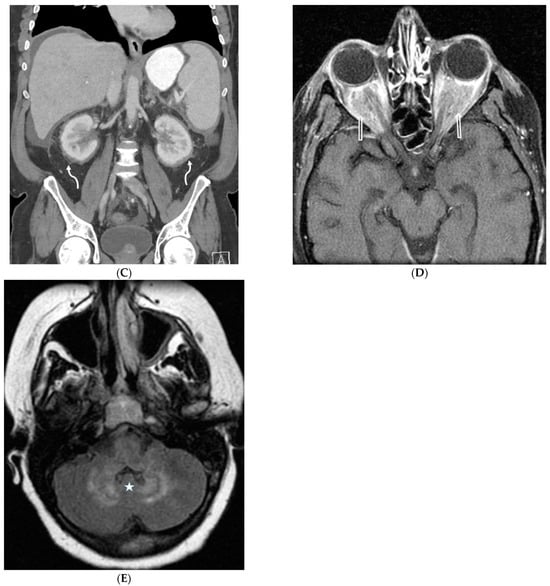
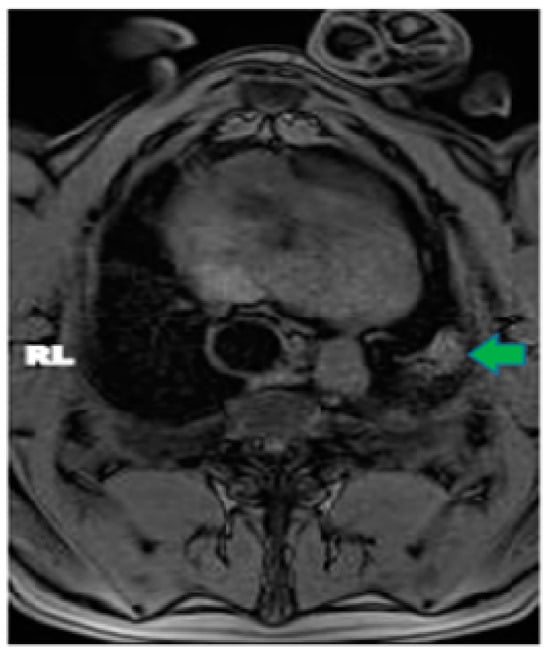
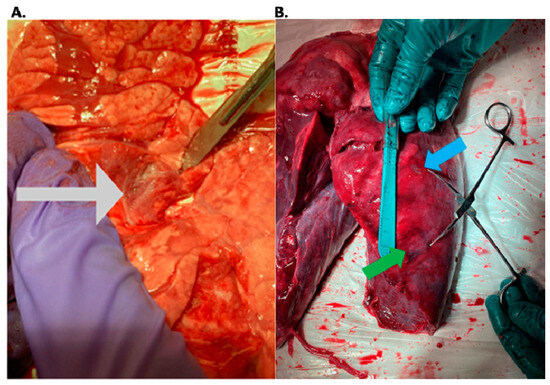
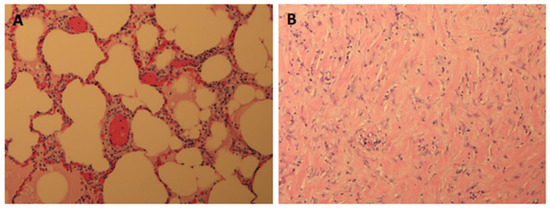
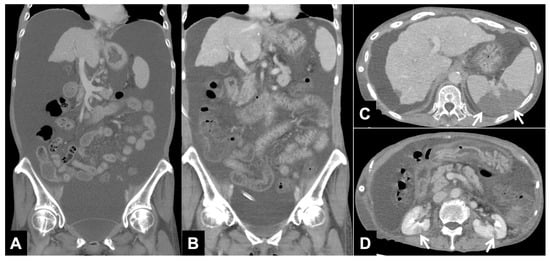
The aim of this study was to evaluate the findings of CT scans in patients with pathologically confirmed primary colorectal squamous-cell carcinoma (SCC). The clinical presentation and CT findings in eight patients with pathologically confirmed primary colorectal squamous-cell carcinoma were retrospectively reviewed by two gastrointestinal radiologists. Hematochezia was the most common symptom (n = 5). The tumors were located in the rectum (n = 7) and sigmoid colon (n = 1). The tumors showed circumferential wall thickening (n = 4), bulky mass (n = 3), or eccentric wall thickening (n = 1). The mean maximal wall thickness of the involved segment was 29.1 mm ± 13.4 mm. The degree of tumoral enhancement observed via CT was well enhanced (n = 4) or moderately enhanced (n = 4). Necrosis within the tumor was found in five patients. The mean total number of metastatic lymph nodes was 3.1 ± 3.3, and the mean short diameter of the largest metastatic lymph node was 16.6 ± 5.7 mm. Necrosis within the metastatic node was observed in six patients. Invasions to adjacent organs were identified in five patients (62.5%). Distant metastasis was detected in only one patient. In summary, primary SCCs that arise from the colorectum commonly present as marked invasive wall thickening or a bulky mass with heterogeneous well-defined enhancement, internal necrosis, and large metastatic lymphadenopathies.
Full article
(This article belongs to the Special Issue Imaging in Cancer Diagnosis)
►
Show Figures

Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}